pyHLAMSA: Python Utility for Star Alleles Sequence Alignment
pyHLAMSA is a Python tool for handling Multiple Sequence Alignments (MSA) of genes with star alleles, specifically focusing on IMGT-HLA, IMGT-KIR, and CYP datasets. Key features include:
- MSA Download: Fetch MSAs for latest/specific versions.
- Gene and Allele Management: Add, delete, list, and select genes and alleles.
- Intron/Exon Operations: Manipulate specific intron/exon segments.
- Position and Region Selection: Choose targeted positions or regions.
- Alignment Operations: Add, view, concatenate, crop, and find differences.
- Genomic and Nucleotide Merging: Seamlessly merge sequences.
- Variant Operations: Calculate consensus, gather statistics, and perform variant-related tasks.
- Biopython Compatibility: Transform alignments into Biopython MultipleSeqAlignment.
- Format Conversion: Load and save MSAs in various formats (VCF, MSF, TXT, BAM, GFF).
pyHLAMSA streamlines complex gene sequence analysis, offering efficient tools for researchers.
Run in command line
You can simply use this package by command line.
If you want to use more powerful function, try the APIs written in below sections.
pip3 install git+https://github.com/linnil1/pyHLAMSA
# show help
pyhlamsa -h
# download kir
pyhlamsa download --family kir --db-folder tmpdir/tmp_kir_db --version 2100 tmpdir/kir --include-genes KIR2DL1 KIR2DL2
# view the msa
pyhlamsa view tmpdir/kir.KIR2DL1 --position 3-100 --include-alleles KIR2DL1*consensus KIR2DL1*063
# save the intron1+exon1 region to kir1.*
pyhlamsa view tmpdir/kir.KIR2DL1 --region intron1 exon1 --name tmpdir/kir1 --save --bam --gff --vcf --fasta-gapless --fasta-msa
Features
1. Read sequences from database in one line
HLAmsa provided a simple way to read HLA data
It can automatically download and read the sequences
>>> from pyhlamsa import HLAmsa
>>> hla = HLAmsa(["A", "B"], filetype="gen",
version="3470")
>>> print(hla.list_genes())
['A', 'B']
>>> print(hla["A"])
<A gen alleles=4101 block=5UTR(301) exon1(80) intron1(130) exon2(335) intron2(273) exon3(413) intron3(666) exon4(287) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)>
KIRmsa can also read sequences of KIR
>>> from pyhlamsa import KIRmsa
# If don't specific the genes, it will read all genes.
>>> kir = KIRmsa(ipd_folder="KIR_v2100", version="2100")
>>> print(kir.list_genes())
['KIR2DL1', 'KIR2DL2', 'KIR2DL3', 'KIR2DL4', 'KIR2DL5', 'KIR2DP1', 'KIR2DS1', 'KIR2DS2', 'KIR2DS3', 'KIR2DS4', 'KIR2DS5', 'KIR3DL1', 'KIR3DL2', 'KIR3DL3', 'KIR3DP1', 'KIR3DS1']
>>> print(kir["KIR2DL1"])
<KIR2DL1 gen alleles=173 block=5UTR(268) exon1(34) intron1(964) exon2(36) intron2(728) exon3(282) intron3(1441) exon4(300) intron4(1534) exon5(294) intron5(3157) exon6(51) intron6(4270) exon7(102) intron7(462) exon8(53) intron8(98) exon9(177) 3UTR(510)>
Kind note: In our modules, exon is actually CDS, so it doesn't include UTR.
2. Merge gene and nuc MSA is quiet simple
This main features give us a chance to use genomic MSA and nucleotide MSA at the same time.
The nucleotide MSA is a exon-only sequence, thus we fill the intron with E after merged.
# merge gen and nuc sequences when loading
>>> hla = HLAmsa(["A"], filetype=["gen", "nuc"],
imgt_alignment_folder="alignment_v3470")
>>> print(hla["A"])
<A gen alleles=7349 block=5UTR(301) exon1(80) intron1(130) exon2(351) intron2(273) exon3(436) intron3(666) exon4(361) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)>
# or manually
>>> a_gen = HLAmsa("A", filetype="gen",
>>> imgt_alignment_folder="alignment_v3470")["A"]
>>> print(a_gen)
<A gen alleles=4101 block=5UTR(301) exon1(80) intron1(130) exon2(335) intron2(273) exon3(413) intron3(666) exon4(287) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)>
>>> a_nuc = HLAmsa("A", filetype="nuc",
>>> imgt_alignment_folder="alignment_v3470")["A"]
>>> print(a_nuc)
<A nuc alleles=7353 block=exon1(80) exon2(351) exon3(436) exon4(361) exon5(117) exon6(33) exon7(48) exon8(5)>
>>> a_gen = a_gen.remove('A*03:437Q')
>>> print(a_gen.merge_exon(a_nuc))
<A gen alleles=7349 block=5UTR(301) exon1(80) intron1(130) exon2(351) intron2(273) exon3(436) intron3(666) exon4(361) intron4(119) exon5(117) intron5(444) exon6(33) intron6(143) exon7(48) intron7(170) exon8(5) 3UTR(302)>
3. Block-based selection: You can select ANY intron/exon.
# select exon2 and exon3
>>> exon23 = a_gen.select_exon([2,3]) # 1-base
>>> print(exon23)
<A nuc alleles=4100 block=exon2(335) exon3(413)>
# select exon2 + intron2 + exon3
>>> e2i2e3 = a_gen.select_block([3,4,5]) # 0-base
>>> print(e2i2e3)
<A alleles=4100 block=exon2(335) intron2(273) exon3(413)>
4. Easy to compare alleles: select and print
# select first 10 alleles
>> exon23_10 = exon23.select_allele(exon23.get_sequence_names()[:10])
>>> print(exon23_10)
<A nuc alleles=10 block=exon2(335) exon3(413)>
# print it
>>> exon23_10.print_alignment()
812 1136
| |
A*01:01:01:01 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:02N ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:03 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:04 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:05 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:06 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:07 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:08 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:09 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
A*01:01:01:10 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC-ATCCAGA TAATGTATGG CTGCGACG-- ----------
# using regex to select
# "|" indicate the border of block, in this case, it's the border of exon2 and exon3
>>> exon23_1field = exon23.select_allele(r"A\*.*:01:01:01$")
>>> exon23_1field.print_alignment_diff()
812 1136
| |
A*01:01:01:01 ACCGAGCGAA CCTGGGGACC CTGCGCGGCT ACTACAACCA GAGCGAGGAC G| GTTCTCACA CC.ATCCAGA TAATGTATGG CTGCGACG.. ..........
A*02:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.G------ GG-------- --------.. ..........
A*03:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- ---------- --------.. ..........
A*11:01:01:01 ------T-G- ---------- ---------- ---------- ---------- -| --------- --.------- ---------- --------.. ..........
A*23:01:01:01 ------A--- ----C---T- GC--T-C--- ---------- --------C- -| --------- --.C------ -G----T--- --------.. ..........
A*25:01:01:01 ------A--G ----C---T- GC--T-C--- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*26:01:01:01 ---------- ---------- ---------- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*29:01:01:01 ---------- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- ----C---.. ..........
A*30:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- ---------- --------.. ..........
A*32:01:01:01 ------A--G ----C---T- GC--T-C--- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*33:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*34:01:01:01 ------T-G- ---------- ---------- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*36:01:01:01 ---------- ---------- ---------- ---------- ---------- -| --------- --.------- ---------- --------.. ..........
A*66:01:01:01 ------T-G- ---------- ---------- ---------- ---------- -| --------- --.------- GG-------- --------.. ..........
A*68:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*69:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.G------ GG-------- --------.. ..........
A*74:01:01:01 ------T-G- ---------- ---------- ---------- --------C- -| --------- --.------- -G-------- --------.. ..........
A*80:01:01:01 ---------- ---------- ---------- ---------- ---------- -| --------- --.------- ---------- --------.. ..........
# show only variantiation
>>> exon23_1field.print_snv()
Total variantion: 71
536 537 541 565 567 570 593 640 654 658 684 721
| | | | | | | | | | | |
A*01:01:01:01 | ATTTCT TCAC ATCCG| CCGGC CG CGG GGA.G | ATCGCCGTGG | .G.ACACG.C | CGTGCGGTTC GACA| AGA..A GATG| CGGGCG CCGT|
A*02:01:01:01 | ------ ---- -----| ----- -- --- ---.- | -----A---- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*03:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*11:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*23:01:01:01 | ------ C--- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*25:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*26:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*29:01:01:01 | -----A C--- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------T ----| ---..G ----| -----A ----|
A*30:01:01:01 | ------ C--- -----| ----- A- T-- A--.- | -----A---- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*32:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------T ----| ---..G ----| ------ ----|
A*33:01:01:01 | -----A C--- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*34:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*36:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..- ----| ------ ----|
A*66:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*68:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*69:01:01:01 | ------ A--- C----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------- ----| ---..G ----| ------ ----|
A*74:01:01:01 | ------ ---- -----| ----- -- --- ---.- | ---------- | .-.-----.- | ---------T ----| ---..G ----| ------ ----|
A*80:01:01:01 | ------ ---- -----| ----- -- --- ---.- | -----A---- | .-.--T--.- | -----A---- ----| ---..G ----| ------ ----|
5. Some useful operation
# Calculate variant frequency of ( A T C G - ) per base
>>> print(exon23.calculate_frequency()[:10])
[[2, 3, 0, 4095, 0], [2, 1, 4097, 0, 0], [0, 4098, 2, 0, 0], [0, 3, 4095, 2, 0], [0, 911, 3188, 0, 1], [0, 1, 4097, 1, 1], [0, 0, 1, 0, 4099], [4097, 0, 0, 2, 1], [4, 3, 4090, 3, 0], [1, 4097, 1, 1, 0]]
# Get consensus(The largest one in frequency) among the msa
>>> exon23.get_consensus(include_gap=True)
'GCTCCC-ACTCCATGAGGTATTTCTTCACATCCGTGTCCCGGCCCGGCCGCGGGGA----GCCCCGCTTCATCGCCGTGGGC-----------------------TACGTGGACG-ACACG-CAGTTCGTGCGGTTCGACAGCGACGCCGCGAGCCAGAGGATGGAGCCG--------------------CGGGCGCCGTGGATA-GAGCAGGAGGGGCCGGAGTATTGGGACCAGGAGACACGGA-------------A-TGTGAAGGCCCACTCACAGACTGACCGAGTGGACCTGGGGACCCTGCGCGGCTACTACAACCAGAGCGAGGCCGGTTCTCACACC-ATCCAGATGATGTATGGCTGCGACG--------------TGGGG-TCGGACGGGCGCTTCCTCCGCGGGTACCAGCA---GGACGCCTACGACGGCAAGGATTAC---ATCGCCCTGAAC------------------------GAGGACCTGCGCTCTTGGACCGCGGCGGAC--------ATGGCGGCTCAGATCACCAAGCGC-AAGT----GGGAGG--CGGCCC-ATGT------------------------------------------GGCGG-AGCAGTTGAGAGCCTACCTGGAGGGCACG--------TGCGTG----GAGTGGCTCCG--CAGATA-CCTGGAGAACGGGAAGGAGACGCTGCAGC-----------------GCACGG'
# Add sequence into MSA
# include_gap=False in get_consensus will ignore the frequency of gap. i.e. choose one of ATCG
>>> a_merged = hla["A"]
>>> consensus_seq = a_merged.get_consensus(include_gap=False)
>>> a_merged.append("A*consensus", consensus_seq)
>>> a_merged.fill_imcomplete("A*consensus")
# Shrink: remove gap if all bases in the column are gap
>>> exon23_10.shrink().print_snv()
gDNA 200
|
A*01:01:01:01 AAGGCCCACT CACAGACTGA CCGAGCGAAC CTGGGGACCC TGCGCGGCTA CTACAACCAG AGCGAGGACG| GTTCTCACAC CATCCAGATA ATGTATGGCT
A*01:01:01:02N ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:03 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:04 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:05 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:06 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:07 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:08 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:09 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
A*01:01:01:10 ---------- ---------- ---------- ---------- ---------- ---------- ----------| ---------- ---------- ----------
# select specific column
>>> a_gen[12:100]
<A alleles=4100 block=(88)>
>>> a_gen[[12,100]].print_alignment_diff()
gDNA 0
|
A*01:01:01:01 GG
A*01:01:01:02N -G
A*01:01:01:03 GG
A*01:01:01:04 --
A*01:01:01:05 --
A*01:01:01:06 --
A*01:01:01:07 --
A*01:01:01:08 --
A*01:01:01:09 --
A*01:01:01:10 --
A*01:01:01:11 GG
A*01:01:01:12 GG
# concat
>>> print(a_gen.select_exon([2]) + a_gen.select_exon([3]))
<A nuc alleles=4100 block=exon2(335) exon3(413)>
6. Write and export the MSA
Causion: If you run merge_exon, or the msa is from filetype=['gen', 'nuc'],
You should fill the E BEFORE save it.
You can fill it by consensus_seq shown before.
-
MultipleSeqAlignment
Transfer to MultipleSeqAlignment
>>> print(a_gen.to_MultipleSeqAlignment()) Alignment with 4100 rows and 3866 columns CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:01 --------------------------------------------...--- A*01:01:01:02N CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:03 --------------------------------------------...--- A*01:01:01:04 --------------------------------------------...AAA A*01:01:01:05 --------------------------------------------...--- A*01:01:01:06 --------------------------------------------...AAA A*01:01:01:07 --------------------------------------------...--- A*01:01:01:08 --------------------------------------------...AAA A*01:01:01:09 --------------------------------------------...--- A*01:01:01:10 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...--- A*01:01:01:11 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:12 --------------------------------------------...AAA A*01:01:01:13 --------------------------------------------...AAA A*01:01:01:14 --------------------------------------------...--- A*01:01:01:15 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...--- A*01:01:01:16 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...--- A*01:01:01:17 CAGGAGCAGAGGGGTCAGGGCGAAGTCCCAGGGCCCCAGGCGTG...AAA A*01:01:01:18 ... --------------------------------------------...--- A*80:07 -
list of SeqRecord
# Save as MSA SeqIO.write(a_gen.to_records(gap=True), "filename.msa.fa", "fasta") # Save as no-gapped sequences SeqIO.write(a_gen.to_records(gap=False), "filename.fa", "fasta") -
fasta
a_gen.to_fasta("filename.msa.fa", gap=True) a_gen.to_fasta("filename.fa", gap=False) -
bam
a_gen.to_bam("filename.bam") -
gff
a_gen.to_gff("filename.gff")After save the MSA as bam and gff, you can show the alignments on IGV
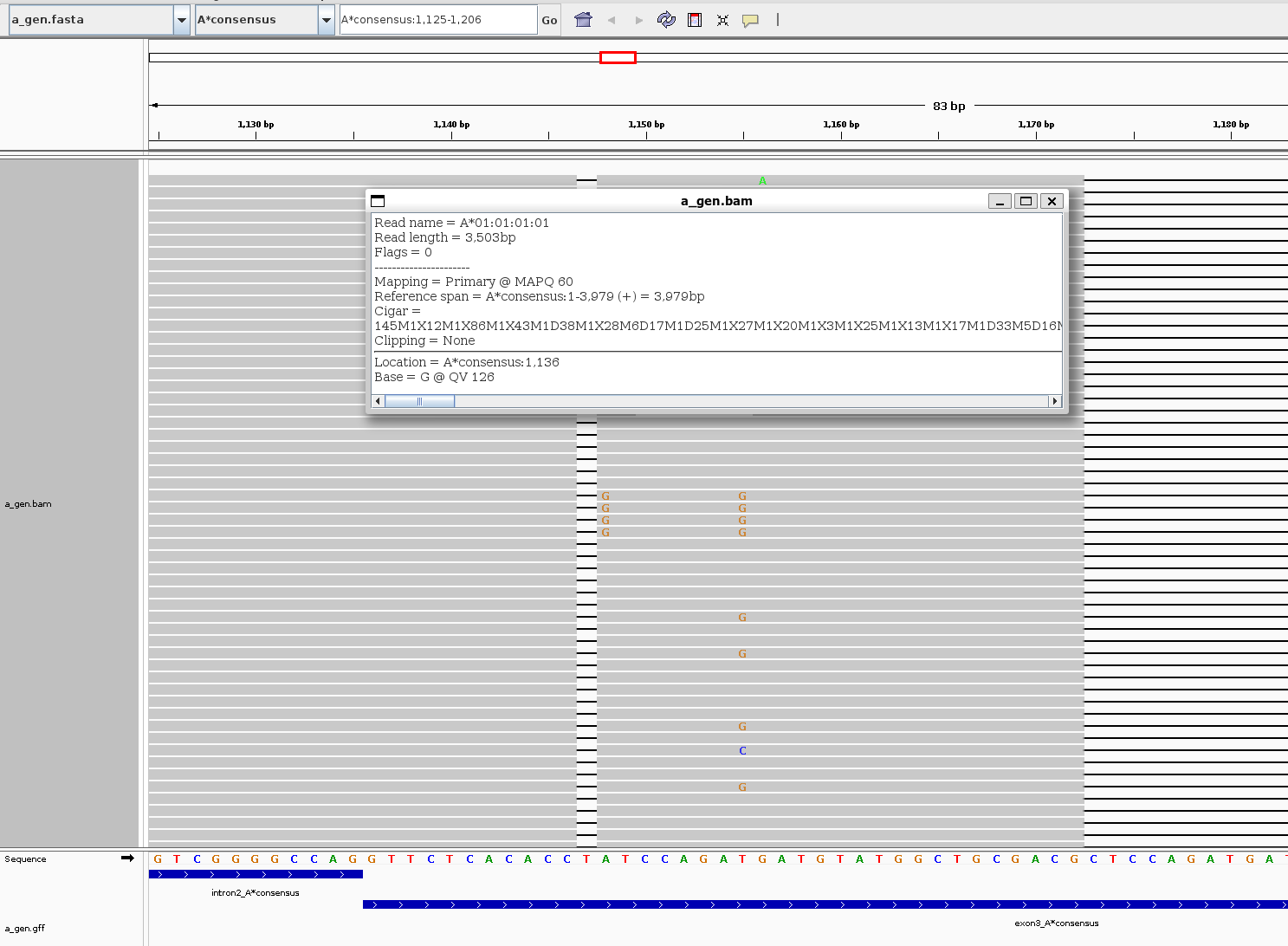
-
vcf
a_gen.to_vcf("filename.vcf.gz") -
IMGT MSA format (xx_gen.txt)
a_gen.to_imgt_alignment("A_gen.txt") a_gen.to_imgt_alignment("A_nuc.txt", seq_type="nuc") -
save/load
Save our model in fasta and json, where json contains block, index information
a_gen.save_msa("a_gen.fa", "a_gen.json") a_gen = Genemsa.load_msa("a_gen.fa", "a_gen.json") -
load msa from other format
pyHLAMSA only support reading from
MultipleSeqAlignment, which is very useful object, can be generate by reading MSA byBio.AlignIO.Checkout https://biopython.org/wiki/AlignIO#file-formats for format supporting.
For example
from pyhlamsa import Genemsa msa = Genemsa.from_MultipleSeqAlignment(AlignIO.read(your_data_path, your_data_format))
TODO
- [x] Testing
- [x] Main function
- [x] exon-only sequence handling
- [x] Reading from file
- [x] Some useful function:
copy,remove,get_sequence_names,__len__,size,split,concat - [x] Cannot handle pseudo exon
- [x] merge blocks and labels
- [x] Fix KIR when merge gen and nuc
- [x] Use index to trace orignal position
- [x] CYP
- [x] Set reference
- [x] Download latest version of IMGT or IPD
- [x] Remove seqtype
- [x] Split code
- [x] save to VCF
- [x] Add command line usage
- [x] Remove msaio
- [x] Rewrite gene Mixins (Too complicated)
- [x] Fix selction and removeing allele by regex
- [x] Change to ACGT order
- [x] Rename: split -> split_block, remove -> remove_allele
- [ ] CDS != exon, (rename it?)
- [ ] Rename: pyHLAMSA -> py_star_msa (MAYBE, becuase the project is originally written for HLA)
Requirement
- python3.9
- biopython
- pysam
- wget
- git
Installation
pip3 install git+https://github.com/linnil1/pyHLAMSA
# or
git clone https://github.com/linnil1/pyHLAMSA
pip3 install -e pyHLAMSA
Appendix: CYP
Steps:
- Download fasta from https://www.pharmvar.org/download
- unzip to
./pharmvar-5.1.10 - Read it by pyhlamsa
# 4. Read it from pyhlamsa import CYPmsa cyp = CYPmsa(pharmvar_folder="./pharmvar-5.1.10") # 5. Test it >>> print(cyp['CYP26A1'].format_variantion_base()) 6407 6448 8142 | | | CYP26A1*1.001 GCGAGCGCGG | ATGTTCCGAA | TCGGGTGTGT CYP26A1*3.001 ---------- | -----A---- | ---------- CYP26A1*2.001 -----A---- | ---------- | ---------- CYP26A1*4.001 ---------- | ---------- | -----C----
Setup Document
pip3 install mkdocs-material mkdocstrings[python-lagacy]==0.18
mkdocs serve
I use python-lagacy 0.18 because inherited_members is not support now
Test
pip3 install -e .
pip3 install pytest black mypy
pytest
black pyhlamsa
mypy pyhlamsa
Tested Version
- CYP: 5.2.2
- HLA: 3.49.0
- KIR: 2.10.0 (The DB in 2.11.0 contains bugs in KIR2DL4/5)
Some QAs
Why not inherit Bio.AlignIO.MultipleSeqAlignment?
The class does't support lot of functions than I expected.
And it's tidious to overwrite most of the functions to maintain our blocks information
Why not use numpy to save the sequence?
Performance issue is not my bottle-neck yet.
Citations
- HLA Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE: IPD-IMGT/HLA Database. Nucleic Acids Research (2020), 48:D948-55
- IMGT Robinson J, Barker DJ, Georgiou X, Cooper MA, Flicek P, Marsh SGE IPD-IMGT/HLA Database Nucleic Acids Research (2020) 48:D948-55
- CYP(PharmVar) Pharmacogene Variation Consortium (PharmVar) at www.PharmVar.org using the following references: Gaedigk et al. 2018, CPT 103(3):399-401 (PMID 29134625); Gaedigk et al. 2020, CPT 107(1):43-46 (PMID 31758698) and Gaedigk et al. 2021, CPT 110(3):542-545 (PMID 34091888)
- This github
Document
See https://linnil1.github.io/pyHLAMSA
Gene Module.
All the msa operation are merged into Genemsa
Genemsa (GenemsaIO, GenemsaTextOp, GenemsaBase)
Genemsa: core object in pyhlamsa
More details written in GenemsaBase
Basic usage:
from pyhlamsa import Genemsa
msa = Genemsa("test1")
msa.set_blocks([3,7])
msa.assume_label("other")
msa.append("A", "GGAGGA--CA")
msa.append("B", "GGGGGAA--A")
msa.print_alignment()
1 4 9
| | |
A GGA|GGA--|CA
B GGG|GGAA-|-A
Source code in pyhlamsa/gene/genemsa.py
class Genemsa(
GenemsaIO,
GenemsaTextOp,
GenemsaBase,
):
"""
Genemsa: core object in pyhlamsa
More details written in GenemsaBase
Basic usage:
``` python
from pyhlamsa import Genemsa
msa = Genemsa("test1")
msa.set_blocks([3,7])
msa.assume_label("other")
msa.append("A", "GGAGGA--CA")
msa.append("B", "GGGGGAA--A")
msa.print_alignment()
1 4 9
| | |
A GGA|GGA--|CA
B GGG|GGAA-|-A
```
"""
__add__(self: ~GenemsaType, msa: ~GenemsaType) -> ~GenemsaType
inherited
special
Source code in pyhlamsa/gene/genemsa.py
def __add__(self: GenemsaType, msa: GenemsaType) -> GenemsaType:
"""
Concat 2 MSA
Example:
>>> print(a_gen.select_exon([2]) + a_gen.select_exon([3]))
<A nuc alleles=4100 block=exon2(335) exon3(413)>
"""
names0 = set(self.get_sequence_names())
names1 = set(msa.get_sequence_names())
if names0 != names1:
raise ValueError(
"Can not concat because some allele is miss: "
+ str(names0.symmetric_difference(names1))
)
new_msa = self.copy()
new_msa.blocks.extend(copy.deepcopy(msa.blocks))
new_msa.index.extend(copy.deepcopy(msa.index))
for name, seq in msa.items():
new_msa.alleles[name] += seq
return new_msa
__getitem__(self: ~GenemsaType, index: Any = None) -> ~GenemsaType
inherited
special
Source code in pyhlamsa/gene/genemsa.py
def __getitem__(self: GenemsaType, index: Any = None) -> GenemsaType:
"""
Extract the region of the sequences by index (start from 0),
but block information will not preserved
Example:
>>> msa = Genemsa("A", "gen")
>>> msa.read_alignment_file("A_gen.txt")
>>> # Inspect 50-100bp in the MSA
>>> extract_msa = msa[50:100]
>>> print(extract_msa)
>>> # Inspect 2nd 3rd 5th bp in the MSA
>>> extract_msa = msa[[1,2,4]]
>>> print(extract_msa)
"""
if not index:
return self.copy()
if not self:
raise ValueError("MSA is empty")
# Extract specific region in alignment
if isinstance(index, int):
index = [index]
if isinstance(index, slice):
alleles = {allele: seq[index] for allele, seq in self.items()}
index = self.index[index]
elif isinstance(index, (tuple, list)):
alleles = {
allele: "".join([seq[i] for i in index]) for allele, seq in self.items()
}
index = [self.index[i] for i in index]
# Fail
else:
raise TypeError("Bad usage")
new_msa = self.copy(copy_allele=False)
new_msa.set_blocks([len(next(iter(alleles.values())))])
new_msa.index = copy.deepcopy(index)
new_msa.extend(alleles)
return new_msa
__init__(self, gene_name: str = 'Unamed', alleles: dict = {}, blocks: Iterable = [], index: Iterable = [], reference: str = '')
inherited
special
Source code in pyhlamsa/gene/genemsa.py
def __init__(
self,
gene_name: str = "Unamed",
alleles: dict[str, str] = {},
blocks: Iterable[BlockInfo] = [],
index: Iterable[IndexInfo] = [],
reference: str = "",
):
"""
Attributes:
gene_name: The name of the gene
alleles: MSA data.
Allele name as the key and the sequence string as the value.
blocks: list of block information
index: list of index(position) information
reference: allele of the msa (Optional)
"""
self.gene_name = gene_name
self.alleles: dict[str, str] = {}
self.blocks = copy.deepcopy(list(blocks or [])) # intron exon length
self.index = copy.deepcopy(list(index or []))
self.logger = logging.getLogger(__name__)
self.reference = reference
if alleles:
self.extend(alleles)
__iter__(self) -> Iterator
inherited
special
Source code in pyhlamsa/gene/genemsa.py
def __iter__(self) -> Iterator[str]:
"""Iter allele like iter(dict)"""
return iter(self.alleles)
__len__(self) -> int
inherited
special
Source code in pyhlamsa/gene/genemsa.py
def __len__(self) -> int:
"""
Get the number of alleles in the MSA
Example:
>>> len(a_gen)
4100
"""
return len(self.alleles)
__repr__(self) -> str
inherited
special
Source code in pyhlamsa/gene/genemsa.py
def __repr__(self) -> str:
"""Show name, number of alleles and block infos in this MSA"""
block_info = " ".join([f"{b.name}({b.length})" for b in self.list_blocks()])
return f"<{self.gene_name} alleles={len(self)} block={block_info}>"
append(self: ~GenemsaType, name: str, seq: str) -> ~GenemsaType
inherited
Add a sequence into MSA (inplace)
Make sure the sequence length is same as in MSA
Source code in pyhlamsa/gene/genemsa.py
def append(self: GenemsaType, name: str, seq: str) -> GenemsaType:
"""
Add a sequence into MSA (inplace)
Make sure the sequence length is same as in MSA
"""
if len(seq) == 0: # OK to add 0 length string
self.alleles[name] = seq
return self
if len(seq) != self.get_length():
raise ValueError("Length not match to alignments")
if name in self:
raise ValueError(f"{name} already exist")
self.alleles[name] = seq
return self
assume_label(self: ~GenemsaType, seq_type: str = 'gen') -> ~GenemsaType
inherited
It will automatically generate the block's label
according on seq_type. (inplace)
seq_type: * gen: 5UTR-exon1-intron1-exon2-...-exon9-3UTR * nuc: exon1-exon2-...-exon9 * other: block1-block2-block3-...
!!! block_length If manually assign the block_length, the old block will be cleared.
Source code in pyhlamsa/gene/genemsa.py
def assume_label(self: GenemsaType, seq_type: str = "gen") -> GenemsaType:
"""
It will automatically generate the block's label
according on `seq_type`. (inplace)
seq_type:
* gen: 5UTR-exon1-intron1-exon2-...-exon9-3UTR
* nuc: exon1-exon2-...-exon9
* other: block1-block2-block3-...
block_length:
If manually assign the block_length, the old block will be cleared.
"""
if not self.get_block_length():
raise ValueError("This msa doesn't have any blocks")
if seq_type == "gen":
assert self.get_block_length() % 2 == 1 and self.get_block_length() >= 3
for i, blk in enumerate(self.blocks):
if i == 0:
blk.type = "five_prime_UTR"
blk.name = "5UTR"
elif i == self.get_block_length() - 1:
blk.type = "three_prime_UTR"
blk.name = "3UTR"
elif i % 2:
blk.type = "exon"
blk.name = f"exon{i // 2 + 1}"
else:
blk.type = "intron"
blk.name = f"intron{i // 2}"
elif seq_type == "nuc":
for i, blk in enumerate(self.blocks):
blk.type = "exon"
blk.name = f"exon{i+1}"
else:
for i, blk in enumerate(self.blocks):
blk.type = "gene_fragment"
blk.name = f"block{i+1}"
# inplace to reset the index
self.index = self.reset_index().index
return self
calculate_frequency(self) -> list
inherited
Calculate ATCG and gap frequency of each bp in MSA
Returns:
| Type | Description |
|---|---|
frequency (list[list[int]]) |
Each items contains the number of ATCG and gap. |
Source code in pyhlamsa/gene/genemsa.py
def calculate_frequency(self) -> list[list[int]]:
"""
Calculate ATCG and gap frequency of each bp in MSA
Returns:
frequency (list[list[int]]):
Each items contains the number of ATCG and gap.
"""
freqs = []
for i in zip(*self.alleles.values()):
freqs.append(
[i.count("A"), i.count("C"), i.count("G"), i.count("T"), i.count("-")]
)
return freqs
contains(self, allele: str) -> bool
inherited
Implement in operator
Examples:
>>> msa_a = HLAmsa("A")["A"]
>>> "A*01:01:01:01" in msa_a
True
Source code in pyhlamsa/gene/genemsa.py
def contains(self, allele: str) -> bool:
"""
Implement `in` operator
Example:
>>> msa_a = HLAmsa("A")["A"]
>>> "A*01:01:01:01" in msa_a
True
"""
return allele in self.alleles
copy(self: ~GenemsaType, copy_allele: bool = True) -> ~GenemsaType
inherited
Clone a new MSA.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
copy_allele |
bool |
Copy the sequences as well |
True |
Source code in pyhlamsa/gene/genemsa.py
def copy(self: GenemsaType, copy_allele: bool = True) -> GenemsaType:
"""
Clone a new MSA.
Args:
copy_allele: Copy the sequences as well
"""
# Child's Type
Genemsa = type(self)
new_msa = Genemsa(
self.gene_name,
blocks=self.blocks,
index=self.index,
reference=self.reference,
alleles=self.alleles if copy_allele else {},
)
return new_msa
extend(self: ~GenemsaType, msa: Union[~GenemsaType, dict[str, str]]) -> ~GenemsaType
inherited
Add MSA's alleles into this MSA (inplace)
Source code in pyhlamsa/gene/genemsa.py
def extend(
self: GenemsaType, msa: Union[GenemsaType, dict[str, str]]
) -> GenemsaType:
"""Add MSA's alleles into this MSA (inplace)"""
if isinstance(msa, GenemsaBase):
if [b.length for b in self.list_blocks()] != [
b.length for b in msa.list_blocks()
]:
raise ValueError("Block length is different")
for name, seq in msa.items():
self.append(name, seq)
return self
fill_incomplete(self: ~GenemsaType, ref_allele: str) -> ~GenemsaType
inherited
Fill the E in exon-only sequences with ref_allele sequence (inplace)
Source code in pyhlamsa/gene/genemsa.py
def fill_incomplete(self: GenemsaType, ref_allele: str) -> GenemsaType:
"""Fill the `E` in exon-only sequences with ref_allele sequence (inplace)"""
if ref_allele not in self:
raise KeyError(f"{ref_allele} not found")
ref_seq = self.get(ref_allele)
for allele, seq in self.items():
if "E" in seq:
# replace it
self.alleles[allele] = "".join(
[seq[i] if seq[i] != "E" else ref_seq[i] for i in range(len(seq))]
)
return self
format_alignment(self, **kwargs_format: Any) -> Iterator
inherited
Transfer MSA to string(generator) To the things in step3 and step4.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
kwargs_format |
Any |
|
{} |
Source code in pyhlamsa/gene/genemsa.py
def format_alignment(self, **kwargs_format: Any) -> Iterator[str]:
"""
Transfer MSA to string(generator)
To the things in step3 and step4.
Args:
kwargs_format:
* show_position_set(set[int]): Force to show the position
* max_page_width(int): The max width of each line (Default = 140char)
"""
if not self:
raise ValueError("MSA is empty")
pages = self._format_page([self.index], **kwargs_format)
yield from chain.from_iterable(map(self._apply_page, pages))
get(self, allele: str) -> str
inherited
Get the sequence by allele name
Source code in pyhlamsa/gene/genemsa.py
def get(self, allele: str) -> str:
"""Get the sequence by allele name"""
return self.alleles[allele]
get_allele_or_error(self, allele: str = '') -> tuple
inherited
Get the sequence by allele name
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
allele |
str |
Allele name. If not provided, reference allele are used |
'' |
Returns:
| Type | Description |
|---|---|
allele_name (str) and its sequence (str) |
Source code in pyhlamsa/gene/genemsa.py
def get_allele_or_error(self, allele: str = "") -> tuple[str, str]:
"""
Get the sequence by allele name
Args:
allele (str): Allele name. If not provided, reference allele are used
Returns:
allele_name (str) and its sequence (str):
"""
if not allele:
allele = self.get_reference()[0]
if allele not in self:
raise ValueError(f"{allele} not found")
return allele, self.get(allele)
get_block(self, block: Union[int, str, pyhlamsa.gene.base.BlockInfo]) -> BlockInfo
inherited
Get block by str or id
Source code in pyhlamsa/gene/genemsa.py
def get_block(self, block: BlockInput) -> BlockInfo:
"""Get block by str or id"""
return self.blocks[self.get_block_index(block)]
get_block_index(self, block: Union[int, str, pyhlamsa.gene.base.BlockInfo]) -> int
inherited
Find the index of the block
Source code in pyhlamsa/gene/genemsa.py
def get_block_index(self, block: BlockInput) -> int:
"""Find the index of the block"""
if isinstance(block, str):
for i, b in enumerate(self.list_blocks()):
if b.name == block:
return i
elif isinstance(block, BlockInfo):
for i, b in enumerate(self.list_blocks()):
if b.name == block.name:
return i
elif isinstance(block, int):
id = block
if id < 0:
id = self.get_block_length() + id
if 0 <= id < self.get_block_length():
return id
else:
raise NotImplementedError(f"Type of {block} not work now")
raise IndexError(f"{block} not found or out of index")
get_block_interval(self, block: Union[int, str, pyhlamsa.gene.base.BlockInfo]) -> tuple
inherited
Calculate the start(included) and end index (excluded) of the block
Source code in pyhlamsa/gene/genemsa.py
def get_block_interval(self, block: BlockInput) -> tuple[int, int]:
"""Calculate the start(included) and end index (excluded) of the block"""
index = self.get_block_index(block)
start = sum(self.blocks[i].length for i in range(index))
return start, start + self.blocks[index].length
get_block_length(self) -> int
inherited
Return list of blocks
Source code in pyhlamsa/gene/genemsa.py
def get_block_length(self) -> int:
"""Return list of blocks"""
return len(self.list_blocks())
get_cigar(self, target_allele: str, ref_allele: str = '') -> list
inherited
Get the cigar string of target_allele from ref_allele
If ref_allele not set, it will automatically find the reference by get_reference
Returns:
| Type | Description |
|---|---|
cigar(list[op_str, num]) |
The list of operator and number of bases |
Exmaple:
cigar = [(M, 1), (X, 1), (D, 2), (M, 2)]
Source code in pyhlamsa/gene/genemsa.py
def get_cigar(
self, target_allele: str, ref_allele: str = ""
) -> list[tuple[str, int]]:
"""
Get the cigar string of target_allele from ref_allele
If ref_allele not set,
it will automatically find the reference by get_reference
Return:
cigar(list[op_str, num]): The list of operator and number of bases
Exmaple:
`cigar = [(M, 1), (X, 1), (D, 2), (M, 2)]`
"""
if target_allele not in self:
raise KeyError(f"{target_allele} not found")
ref_allele, _ = self.get_allele_or_error(ref_allele)
return cigar.calculate_cigar(self.get(ref_allele), self.get(target_allele))
get_consensus(self, include_gap: bool = False) -> str
inherited
Generate the consensus sequence by choosing maximum frequency base
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
include_gap |
bool |
Allow consensus contains gap if gap is the maximum item. If include_gap=False and all the base on that position is gap (not shrinked before), it will warning and fill with A.
|
False |
Examples:
a0 CCATT-GGT--GTCGGGTTTCCAG
a1 CCACTGGGT--ATCGGGTTTCCAG
c2 CAATTGGGT--GTCGGGT---AAG
consensus CCATTGGGT--GTCGGGTTTCCAG
consensus(no-gap) CCATTGGGTAAGTCGGGTTTCCAG
Source code in pyhlamsa/gene/genemsa.py
def get_consensus(self, include_gap: bool = False) -> str:
"""
Generate the consensus sequence by choosing maximum frequency base
Args:
include_gap (bool):
Allow consensus contains gap if gap is the maximum item.
If include_gap=False and all the base on that position is gap
(not shrinked before),
it will warning and fill with A.
`E` will be ignored.
Example:
```
a0 CCATT-GGT--GTCGGGTTTCCAG
a1 CCACTGGGT--ATCGGGTTTCCAG
c2 CAATTGGGT--GTCGGGT---AAG
consensus CCATTGGGT--GTCGGGTTTCCAG
consensus(no-gap) CCATTGGGTAAGTCGGGTTTCCAG
```
"""
freqs = self.calculate_frequency()
if not include_gap:
if any(sum(f[:4]) == 0 for f in freqs):
self.logger.warning(
"MSA contains gap, try .shrink() before .get_consensus()"
)
max_ind = [max(range(4), key=lambda i: f[i]) for f in freqs]
else:
max_ind = [max(range(5), key=lambda i: f[i]) for f in freqs]
return "".join(map(lambda i: "ACGT-"[i], max_ind))
get_length(self) -> int
inherited
Get the length of MSA
Source code in pyhlamsa/gene/genemsa.py
def get_length(self) -> int:
"""Get the length of MSA"""
# 0 sequences is allow
if not self:
# another way to calculate length
return sum(i.length for i in self.list_blocks())
else:
# when extend, reference allele may not exists here
# return len(self.get_reference()[1])
return len(next(iter(self.items()))[1])
get_reference(self) -> tuple
inherited
Get the reference in MSA, if not existed, output the first one
Returns:
| Type | Description |
|---|---|
tuple |
(allele_name, allele_seq) |
Source code in pyhlamsa/gene/genemsa.py
def get_reference(self) -> tuple[str, str]:
"""
Get the reference in MSA, if not existed, output the first one
Returns:
(allele_name, allele_seq)
"""
if self.reference in self:
return (self.reference, self.get(self.reference))
if not self:
raise ValueError("MSA is empty")
self.logger.warning(
f"Reference {self.reference} not existed in MSA."
" Using the first allele instead."
)
allele, seq = next(iter(self.items()))
self.reference = allele # set it
return allele, seq
get_sequence_names(self) -> KeysView
inherited
Same as list_alleles
Source code in pyhlamsa/gene/genemsa.py
def get_sequence_names(self) -> KeysView[str]:
"""Same as list_alleles"""
return self.list_alleles()
get_variantion_base(self) -> list
inherited
Get the base positions where variation occurs
Examples:
msa:
s0: AAT
s1: AAC
s2: CAC
>>> msa.get_variantion_base()
[0, 2]
Returns:
| Type | Description |
|---|---|
positions |
Each integer represent the position of variation |
Source code in pyhlamsa/gene/genemsa.py
def get_variantion_base(self) -> list[int]:
"""
Get the base positions where variation occurs
Example:
```
msa:
s0: AAT
s1: AAC
s2: CAC
>>> msa.get_variantion_base()
[0, 2]
```
Returns:
positions:
Each integer represent the position of variation
"""
freqs = self.calculate_frequency()
num = len(self)
base = []
for i, freq in enumerate(freqs):
if num not in freq:
base.append(i)
return base
items(self) -> Iterable
inherited
list allele name along with allele sequence like dict.items()
Source code in pyhlamsa/gene/genemsa.py
def items(self) -> Iterable[tuple[str, str]]:
"""list allele name along with allele sequence like dict.items()"""
return self.alleles.items()
list_alleles(self) -> KeysView
inherited
List all the allele's sequence name in MSA
Examples:
>>> a_gen.list_alleles()[:3]
['A*01:01:01:01', 'A*01:01:01:02N', 'A*01:01:01:03']
Source code in pyhlamsa/gene/genemsa.py
def list_alleles(self) -> KeysView[str]:
"""
List all the allele's sequence name in MSA
Example:
>>> a_gen.list_alleles()[:3]
['A*01:01:01:01', 'A*01:01:01:02N', 'A*01:01:01:03']
"""
return self.alleles.keys()
list_blocks(self) -> list
inherited
Return list of blocks
Source code in pyhlamsa/gene/genemsa.py
def list_blocks(self) -> list[BlockInfo]:
"""Return list of blocks"""
return self.blocks
list_index(self) -> list
inherited
Return list of index
Source code in pyhlamsa/gene/genemsa.py
def list_index(self) -> list[IndexInfo]:
"""Return list of index"""
return self.index
merge_exon(self: ~GenemsaType, msa_nuc: ~GenemsaType) -> ~GenemsaType
inherited
Merge nuc MSA into gen MSA
It's allow that nuc MSA has new allele name than gen MSA,
Genemsa will add the sequence in MSA, and the intron will fill by E
If the exon part of gen MSA is differnet (e.g. less gapped) from nuc MSA, Genemsa will try to merge if it can
Note that the index will be reset
Examples:
# case1
msa_gen:
1: "AA|TT|CC",
2: "AA|TC|CC",
msa_nuc:
3: "TC",
After merge:
1: "AA|TT|CC",
2: "AA|TC|CC",
3: "EE|TC|EE"
# case2
msa_gen:
1: "AA|TT|CC",
2: "AA|TC|CC",
msa_nuc:
1: "TT-",
2: "T-C",
4: "TTC",
After merge:
1: "AA|TT-|CC",
2: "AA|T-C|CC",
4: "EE|TTC|EE"
Source code in pyhlamsa/gene/genemsa.py
def merge_exon(self: GenemsaType, msa_nuc: GenemsaType) -> GenemsaType:
"""
Merge nuc MSA into gen MSA
It's allow that nuc MSA has new allele name than gen MSA,
Genemsa will add the sequence in MSA, and the intron will fill by `E`
If the exon part of gen MSA is differnet (e.g. less gapped) from nuc MSA,
Genemsa will try to merge if it can
Note that the index will be reset
Example:
```
# case1
msa_gen:
1: "AA|TT|CC",
2: "AA|TC|CC",
msa_nuc:
3: "TC",
After merge:
1: "AA|TT|CC",
2: "AA|TC|CC",
3: "EE|TC|EE"
```
```
# case2
msa_gen:
1: "AA|TT|CC",
2: "AA|TC|CC",
msa_nuc:
1: "TT-",
2: "T-C",
4: "TTC",
After merge:
1: "AA|TT-|CC",
2: "AA|T-C|CC",
4: "EE|TTC|EE"
```
"""
# A mapping from gen name to nuc index
nuc_name_index = {
b.name: i for i, b in enumerate(msa_nuc.list_blocks()) if b.type == "exon"
}
# check it's one-to-one mapping
exon_set = set(b.name for b in self.list_blocks() if b.type == "exon")
if set(nuc_name_index.keys()) != exon_set:
raise ValueError(
f"Cannot match blocks: " f"gen={exon_set} nuc={nuc_name_index.keys()}"
)
# create new msa and make sure the order of alleles
new_msa = self.copy(copy_allele=False)
new_msa.set_blocks([])
new_msa.index = []
new_msa.extend(
{
name: ""
for name in self.get_sequence_names() | msa_nuc.get_sequence_names()
}
)
# allele names
gen_names = set(self.get_sequence_names())
nuc_names = set(msa_nuc.get_sequence_names())
exclude_name: set[str] = set()
# block-wise
msas_gen = self.split_block()
msas_nuc = msa_nuc.split_block()
for i_gen, blk_gen in enumerate(self.blocks):
# intron -> fill with E
if blk_gen.name not in nuc_name_index:
for name in nuc_names - gen_names:
msas_gen[i_gen].append(name, "E" * blk_gen.length)
new_msa += msas_gen[i_gen].remove_allele(
list(exclude_name), inplace=True
)
# exon -> check before merge
else:
i_nuc = nuc_name_index[blk_gen.name]
# content change or length change
if msas_nuc[i_nuc].get_length() != msas_gen[i_gen].get_length() or any(
msas_nuc[i_nuc].get(name) != msas_gen[i_gen].get(name)
for name in (nuc_names & gen_names)
):
# check before merge
if len(gen_names - nuc_names):
raise ValueError(
f"Some alleles doesn't exist in nuc MSA: "
f"{gen_names - nuc_names}"
)
diff_name = filter(
lambda name: msas_nuc[i_nuc].get(name).replace("-", "")
!= msas_gen[i_gen].get(name).replace("-", ""),
gen_names,
)
diff_names = list(diff_name)
if diff_names:
self.logger.warning(
f"Some exon sequences in gen MSA "
f"is not same as in nuc MSA "
f"{blk_gen.name}: {diff_names}"
)
new_msa.remove_allele(diff_names)
exclude_name.update(diff_names)
new_msa += msas_nuc[i_nuc].remove_allele(list(exclude_name))
return new_msa.reset_index()
meta_to_json(self) -> dict[str, Any]
inherited
Extract all meta information about this msa into dict(json)
Source code in pyhlamsa/gene/genemsa.py
def meta_to_json(self) -> dict[str, Any]:
"""Extract all meta information about this msa into dict(json)"""
meta = {
"index": [dataclasses.asdict(i) for i in self.index],
"blocks": [dataclasses.asdict(b) for b in self.blocks],
"name": self.gene_name,
"reference": self.reference,
}
return meta
print_alignment(self, **kwargs_format: Any) -> None
inherited
Print the MSA
Note that the index shown on output header is 1-base absolute position.
Also, the index is absolute index, if you you want continuous positiont,
use .reset_index() first.
*indicate deletionATCGindicate the SNVEindicate the null base in exon-only sequence|is intron and exon boundary
Examples:
>>> a = msa.select_allele(r"A\*.*:01:01:01$").select_exon([6,7])
>>> print(''.join(a.format_alignment()))
# or simply use
>>> a.print_alignment()
3166 3342
| |
A*01:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*02:01:01:01 ATAGAAAAGG AGGGAGCTAC TCTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*03:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*11:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*23:01:01:01 ATAGAAAAGG AGGGAGCTAC TCTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*25:01:01:01 ATAGAAAAGG AGGGAGCTAC TCTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
Source code in pyhlamsa/gene/genemsa.py
def print_alignment(self, **kwargs_format: Any) -> None:
"""
Print the MSA
Note that the index shown on output header is 1-base absolute position.
Also, the index is absolute index, if you you want continuous positiont,
use `.reset_index()` first.
* `*` indicate deletion
* `ATCG` indicate the SNV
* `E` indicate the null base in exon-only sequence
* `|` is intron and exon boundary
Examples:
>>> a = msa.select_allele(r"A\\*.*:01:01:01$").select_exon([6,7])
>>> print(''.join(a.format_alignment()))
# or simply use
>>> a.print_alignment()
3166 3342
| |
A*01:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*02:01:01:01 ATAGAAAAGG AGGGAGCTAC TCTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*03:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*11:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*23:01:01:01 ATAGAAAAGG AGGGAGCTAC TCTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
A*25:01:01:01 ATAGAAAAGG AGGGAGCTAC TCTCAGGCTG CAA| GCAGTGA CAGTGCCCAG
"""
page_str = self.format_alignment(**kwargs_format)
for i in page_str:
print(i, end="")
print_alignment_diff(self, ref_allele: str = '', **kwargs_format: Any) -> None
inherited
Print the sequences of all alleles diff from ref_allele sequence.
Same as print_alignment, but
once the base of alleles is same as the base in reference sequence,
- will be used
Returns:
| Type | Description |
|---|---|
str |
A formatted string |
Examples:
>>> a = msa.select_allele(r"A\*.*:01:01:01$").select_exon([6,7])
>>> print(a.format_alignment_diff())
3166 3342
| |
A*01:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCC
A*02:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*03:01:01:01 ---------- ---------- ---------- ---| ------- -------
A*11:01:01:01 ---------- ---------- ---------- ---| ------- -------
A*23:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*25:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*26:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*29:01:01:01 ---------- ------C--- T--------- ---| ------- -------
Source code in pyhlamsa/gene/genemsa.py
def print_alignment_diff(self, ref_allele: str = "", **kwargs_format: Any) -> None:
"""
Print the sequences of all alleles diff from `ref_allele` sequence.
Same as print_alignment, but
once the base of alleles is same as the base in reference sequence,
`-` will be used
Returns:
str: A formatted string
Examples:
```
>>> a = msa.select_allele(r"A\\*.*:01:01:01$").select_exon([6,7])
>>> print(a.format_alignment_diff())
3166 3342
| |
A*01:01:01:01 ATAGAAAAGG AGGGAGTTAC ACTCAGGCTG CAA| GCAGTGA CAGTGCC
A*02:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*03:01:01:01 ---------- ---------- ---------- ---| ------- -------
A*11:01:01:01 ---------- ---------- ---------- ---| ------- -------
A*23:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*25:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*26:01:01:01 ---------- ------C--- T--------- ---| ------- -------
A*29:01:01:01 ---------- ------C--- T--------- ---| ------- -------
```
"""
new_msa = self._calc_diff_msa(ref_allele)
new_msa.print_alignment(**kwargs_format)
print_alignment_from_center(self: ~GenemsaType, pos: Union[int, collections.abc.Iterable[int]], left: int = 5, right: int = 5, **kwargs_format: Any) -> None
inherited
Print all alleles sequences from the center of specific position
Check .format_alignment() for output format detail
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
pos |
int or list of int |
The (0-base relative) positions |
required |
left |
int |
How many base shall print at the left of the center |
5 |
right |
int |
How many base shall print at the right of the center |
5 |
Returns:
| Type | Description |
|---|---|
str |
A formatted string |
Examples:
gDNA 3022
|
HLA-A*03:02 AGAGAAAAAT
HLA-A*01:40 -----G----
HLA-A*03:20 -----G----
Source code in pyhlamsa/gene/genemsa.py
def print_alignment_from_center(
self: GenemsaType,
pos: Union[int, Iterable[int]],
left: int = 5,
right: int = 5,
**kwargs_format: Any,
) -> None:
"""
Print all alleles sequences from the center of specific position
Check `.format_alignment()` for output format detail
Args:
pos (int or list of int): The (0-base relative) positions
left (int): How many base shall print at the left of the center
right (int): How many base shall print at the right of the center
Returns:
str: A formatted string
Examples:
```
gDNA 3022
|
HLA-A*03:02 AGAGAAAAAT
HLA-A*01:40 -----G----
HLA-A*03:20 -----G----
```
"""
if isinstance(pos, int):
want_pos = [pos]
else:
want_pos = list(pos)
if not want_pos:
return
new_msa = self._calc_center_msa(want_pos, left, right)
show_position_set = set(self.index[i].pos for i in want_pos)
new_msa.print_alignment(show_position_set=show_position_set, **kwargs_format)
print_snv(self, **kwargs_format: Any) -> None
inherited
A handy function to show all the variation between the alleles
Note: the | in the output string is NOT intron and exon boundary
Check .format_alignment() for output format detail
Returns:
| Type | Description |
|---|---|
str |
A formatted string |
Examples:
Total variantion: 71
308 314 329 342 349
| | | | |
A*01:01:01:01 TGGCCGTCAT GGCGCC| CCCT CCTCCT| ACTC TCGGGGGCCC TGG
A*02:01:01:01 ---------- ------| ---- -G----| ---- --------T- ---
A*03:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*11:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*23:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*25:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*26:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*29:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*30:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*32:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*33:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*34:01:01:01 -----A---- ------| ---- -G----| ---- ---------- ---
A*36:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*66:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*68:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*69:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*74:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*80:01:01:01 ---------- -C----| ---- ------| ---- ---------- ---
Source code in pyhlamsa/gene/genemsa.py
def print_snv(self, **kwargs_format: Any) -> None:
"""
A handy function to show all the variation between the alleles
Note: the `|` in the output string is NOT intron and exon boundary
Check `.format_alignment()` for output format detail
Returns:
str: A formatted string
Example:
```
Total variantion: 71
308 314 329 342 349
| | | | |
A*01:01:01:01 TGGCCGTCAT GGCGCC| CCCT CCTCCT| ACTC TCGGGGGCCC TGG
A*02:01:01:01 ---------- ------| ---- -G----| ---- --------T- ---
A*03:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*11:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*23:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*25:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*26:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*29:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*30:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*32:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*33:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*34:01:01:01 -----A---- ------| ---- -G----| ---- ---------- ---
A*36:01:01:01 ---------- ------| ---- ------| ---- ---------- ---
A*66:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*68:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*69:01:01:01 ---------- ------| ---- -G----| ---- ---------- ---
A*74:01:01:01 ---------- ------| ---- ------| ---- -T-------- ---
A*80:01:01:01 ---------- -C----| ---- ------| ---- ---------- ---
```
"""
msa, grouped_bases = self._calc_snp_msa()
show_position_set = set(
self.index[i].pos for bases in grouped_bases for i in bases
)
print(f"#Total variantion: {sum(map(len, grouped_bases))}\n")
msa = msa._calc_diff_msa()
msa.print_alignment(show_position_set=show_position_set, **kwargs_format)
remove_allele(self: ~GenemsaType, query: Union[str, collections.abc.Iterable[str]], inplace: bool = True) -> ~GenemsaType
inherited
Remove allele sequences by regex(when query is string) or by exactly deleting (when query is a list)
Source code in pyhlamsa/gene/genemsa.py
def remove_allele(
self: GenemsaType, query: Union[str, Iterable[str]], inplace: bool = True
) -> GenemsaType:
"""
Remove allele sequences by regex(when query is string)
or by exactly deleting (when query is a list)
"""
if inplace:
new_msa = self
else:
new_msa = self.copy()
if isinstance(query, str):
for allele in list(self):
if re.match(query, allele):
del new_msa.alleles[allele]
elif isinstance(query, Iterable):
for allele in query:
del new_msa.alleles[allele]
else:
raise NotImplementedError
return new_msa
reset_index(self: ~GenemsaType) -> ~GenemsaType
inherited
Reset index: The old position information will be discard.
Each position information will be counted from 0 and the label and name will copy from its block information
Examples:
>>> print(a.format_alignment_diff())
101 103 105 107 109 111 113 115 117 119
| | | | | | | | | |
A*01:01:01:01 G G T C C A C C G A
A*01:01:01:02N - - - - - - - - - -
>>> print(a.reset_index().format_alignment_diff())
1
|
A*01:01:01:01 GGTCCACCGA
A*01:01:01:02N ----------
Source code in pyhlamsa/gene/genemsa.py
def reset_index(self: GenemsaType) -> GenemsaType:
"""
Reset index:
The old position information will be discard.
Each position information will be counted from 0 and
the label and name will copy from its block information
Example:
``` python
>>> print(a.format_alignment_diff())
101 103 105 107 109 111 113 115 117 119
| | | | | | | | | |
A*01:01:01:01 G G T C C A C C G A
A*01:01:01:02N - - - - - - - - - -
>>> print(a.reset_index().format_alignment_diff())
1
|
A*01:01:01:01 GGTCCACCGA
A*01:01:01:02N ----------
```
"""
new_msa = self.copy()
start = 0
new_msa.index = []
for block in self.list_blocks():
for _ in range(block.length):
new_msa.index.append(
IndexInfo(
pos=start,
type=block.type,
name=block.name,
)
)
start += 1
assert start == self.get_length()
return new_msa
reverse_complement(self: ~GenemsaType) -> ~GenemsaType
inherited
Reverse the sequences
Source code in pyhlamsa/gene/genemsa.py
def reverse_complement(self: GenemsaType) -> GenemsaType:
"""Reverse the sequences"""
new_msa = self.copy(copy_allele=False)
new_msa.blocks = copy.deepcopy(list(reversed(self.blocks)))
new_msa.index = copy.deepcopy(list(reversed(self.index)))
new_msa.extend(
{allele: str(Seq(seq).reverse_complement()) for allele, seq in self.items()}
)
return new_msa
save_msa(self, file_fasta: FileType, file_json: FileType) -> None
inherited
Save Genemsa to pyhlamsa format (fasta and json)
Source code in pyhlamsa/gene/genemsa.py
def save_msa(self, file_fasta: FileType, file_json: FileType) -> None:
"""Save Genemsa to pyhlamsa format (fasta and json)"""
self.to_fasta(file_fasta, gap=True)
f_json, require_close = getFileHandle(file_json)
json.dump(self.meta_to_json(), f_json)
if require_close:
f_json.close()
select_allele(self: ~GenemsaType, query: Union[str, collections.abc.Iterable[str]]) -> ~GenemsaType
inherited
Select allele name by regex(when query is string) or by exactly selecting (when query is a list)
Examples:
>>> # select allele name start with A*01:01
>>> msa.select_allele(r"A\*01:01.*")
>>> # select allele by list of string
>>> msa.select_allele(["A*01:01", "A*02:03"])
Source code in pyhlamsa/gene/genemsa.py
def select_allele(
self: GenemsaType, query: Union[str, Iterable[str]]
) -> GenemsaType:
"""
Select allele name by regex(when query is string)
or by exactly selecting (when query is a list)
Examples:
>>> # select allele name start with A*01:01
>>> msa.select_allele(r"A\\*01:01.*")
>>> # select allele by list of string
>>> msa.select_allele(["A*01:01", "A*02:03"])
"""
new_msa = self.copy(copy_allele=False)
if isinstance(query, str):
new_msa.extend(
{allele: seq for allele, seq in self.items() if re.match(query, allele)}
)
elif isinstance(query, Iterable):
new_msa.extend({allele: self.get(allele) for allele in query})
else:
raise NotImplementedError
return new_msa
select_block(self: ~GenemsaType, index: Iterable) -> ~GenemsaType
inherited
Extract blocks by index
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
index |
list of int |
Leave empty if you want all the blocks. Index start from 0. e.g.
or you can use list of string, it will select by block name |
required |
Examples:
>>> a_gen.select_block([-1])
<A alleles=4101 block=3UTR(302)>
>>> a_gen.select_block([2, 3])
<A alleles=4101 block=intron1(130) exon2(335)>
>>> a_gen.select_block(["5UTR", "exon3"])
<A alleles=4101 block=5UTR(301) exon3(413)>
Source code in pyhlamsa/gene/genemsa.py
def select_block(self: GenemsaType, index: Iterable[BlockInput]) -> GenemsaType:
"""
Extract blocks by index
Args:
index (list of int): Leave empty if you want all the blocks.
Index start from 0. e.g.
* 0 for 5-UTR
* 1 for exon1
* 2 for intron1
* 3 for exon2
* 4 for 3-UTR(for two exons gene)
* -1 for last block
or you can use list of string,
it will select by block name
Example:
>>> a_gen.select_block([-1])
<A alleles=4101 block=3UTR(302)>
>>> a_gen.select_block([2, 3])
<A alleles=4101 block=intron1(130) exon2(335)>
>>> a_gen.select_block(["5UTR", "exon3"])
<A alleles=4101 block=5UTR(301) exon3(413)>
"""
new_msa = self.copy(copy_allele=False)
# choose block index by index
new_block = []
new_index = []
all_pos = []
index_ids = [self.get_block_index(i) for i in index]
for i in index_ids:
new_block.append(self.blocks[i])
start, end = self.get_block_interval(i)
all_pos.append((start, end))
new_index.extend(self.index[start:end])
new_msa.blocks = new_block
new_msa.index = new_index
# extract the sequences inside block region
for allele, gen_seq in self.items():
new_seq = "".join([gen_seq[start:end] for start, end in all_pos])
new_msa.append(allele, new_seq)
return new_msa.copy()
select_complete(self: ~GenemsaType) -> ~GenemsaType
inherited
Select non exon-only sequences (No E in the sequence)
Source code in pyhlamsa/gene/genemsa.py
def select_complete(self: GenemsaType) -> GenemsaType:
"""Select non exon-only sequences (No `E` in the sequence)"""
new_msa = self.copy(copy_allele=False)
new_msa.extend({allele: seq for allele, seq in self.items() if "E" not in seq})
return new_msa
select_exon(self: ~GenemsaType, exon_index: Iterable = []) -> ~GenemsaType
inherited
Extract the exon by index.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
exon_index |
list[str|int] |
Index start from 1. i.e.
Leave empty if you want all the exons If the exon_index contains list of string, it will select by name |
[] |
Examples:
>>> a_gen.select_exon([2]))
<A gen alleles=7350 block=exon2(351)>
>>> a_gen.select_exon([2, 3]))
<A nuc alleles=7350 block=exon2(351) exon3(436)>
>>> a_gen.select_exon(["exon2", "exon3"]))
<A nuc alleles=7350 block=exon2(351) exon3(436)>
Source code in pyhlamsa/gene/genemsa.py
def select_exon(
self: GenemsaType, exon_index: Iterable[BlockInput] = []
) -> GenemsaType:
"""
Extract the exon by index.
Args:
exon_index (list[str|int]): Index start from 1. i.e.
* 1 for exon1
* 2 for exon2
Leave empty if you want all the exons
If the exon_index contains list of string,
it will select by name
Example:
>>> a_gen.select_exon([2]))
<A gen alleles=7350 block=exon2(351)>
>>> a_gen.select_exon([2, 3]))
<A nuc alleles=7350 block=exon2(351) exon3(436)>
>>> a_gen.select_exon(["exon2", "exon3"]))
<A nuc alleles=7350 block=exon2(351) exon3(436)>
"""
exons = [b for b in self.list_blocks() if b.type == "exon"]
# If not specific the index, extract all exons
exon_list: list[BlockInput] = []
if not exon_index:
exon_list = exons # type: ignore
else:
for i in exon_index:
if isinstance(i, int):
# exon -> blocks position
if i < 1 or i > len(exons):
raise IndexError(f"{i} is out of exon index")
i = exons[i - 1]
exon_list.append(i)
# check
for i in exon_list:
block = self.get_block(i)
if block.type != "exon":
raise IndexError(f"{block} is not exon: input={i}")
return self.select_block(exon_list)
select_incomplete(self: ~GenemsaType) -> ~GenemsaType
inherited
Select exon-only sequences (E exists in the sequence)
Source code in pyhlamsa/gene/genemsa.py
def select_incomplete(self: GenemsaType) -> GenemsaType:
"""Select exon-only sequences (`E` exists in the sequence)"""
new_msa = self.copy(copy_allele=False)
new_msa.extend({allele: seq for allele, seq in self.items() if "E" in seq})
return new_msa
set_blocks(self: ~GenemsaType, blocks: Iterable) -> ~GenemsaType
inherited
Set blocks (inplace)
Source code in pyhlamsa/gene/genemsa.py
def set_blocks(
self: GenemsaType, blocks: Iterable[Union[int, BlockInfo]]
) -> GenemsaType:
"""Set blocks (inplace)"""
new_blocks = []
for i in blocks:
if isinstance(i, int):
new_blocks.append(BlockInfo(length=i))
else:
new_blocks.append(copy.deepcopy(i))
if len(self) and self.get_length() != sum(blk.length for blk in new_blocks):
raise ValueError("Total block length not match to alignments")
self.blocks = new_blocks
return self
set_reference(self: ~GenemsaType, allele: str) -> ~GenemsaType
inherited
Set the reference in msa (inplace)
Source code in pyhlamsa/gene/genemsa.py
def set_reference(self: GenemsaType, allele: str) -> GenemsaType:
"""Set the reference in msa (inplace)"""
if allele not in self:
self.logger.warning(f"Cannot find {allele} in MSA")
self.reference = allele
return self
shrink(self: ~GenemsaType) -> ~GenemsaType
inherited
Remove empty base if all bases in that column is gap
Source code in pyhlamsa/gene/genemsa.py
def shrink(self: GenemsaType) -> GenemsaType:
"""Remove empty base if all bases in that column is gap"""
# index to delete
freqs = self.calculate_frequency()
masks = [f[4] != sum(f) for f in freqs]
new_msa = self.copy(copy_allele=False)
# recalcuate blocks
for i in range(len(self.blocks)):
start, end = self.get_block_interval(i)
new_msa.blocks[i].length = sum(masks[start:end])
assert sum(masks) == new_msa.get_length()
new_msa.index = [new_msa.index[i] for i in range(len(masks)) if masks[i]]
# remove base in allele
for allele, seq in self.items():
new_msa.append(allele, "".join(seq[i] for i in range(len(seq)) if masks[i]))
return new_msa
size(self) -> tuple
inherited
Get the size (num_of_allele, length_of_sequence)
Source code in pyhlamsa/gene/genemsa.py
def size(self) -> tuple[int, int]:
"""Get the size (num_of_allele, length_of_sequence)"""
return (len(self), self.get_length())
sort(self: ~GenemsaType) -> ~GenemsaType
inherited
Sort the allele by their sequences (inplace)
Source code in pyhlamsa/gene/genemsa.py
def sort(self: GenemsaType) -> GenemsaType:
"""Sort the allele by their sequences (inplace)"""
self.alleles = dict(sorted(self.items(), key=lambda i: i[1]))
return self
sort_name(self: ~GenemsaType) -> ~GenemsaType
inherited
Sort the allele by alelle name (inplace)
Source code in pyhlamsa/gene/genemsa.py
def sort_name(self: GenemsaType) -> GenemsaType:
"""Sort the allele by alelle name (inplace)"""
self.alleles = dict(sorted(self.items(), key=lambda i: i[0]))
return self
split_block(self: ~GenemsaType) -> list
inherited
Split the msa by blocks
Source code in pyhlamsa/gene/genemsa.py
def split_block(self: GenemsaType) -> list[GenemsaType]:
"""Split the msa by blocks"""
return [self.select_block([i]) for i in range(len(self.list_blocks()))]
to_MultipleSeqAlignment(self) -> MultipleSeqAlignment
inherited
Transfer this object to MultipleSeqAlignment(biopython)
Source code in pyhlamsa/gene/genemsa.py
def to_MultipleSeqAlignment(self) -> MultipleSeqAlignment:
"""Transfer this object to MultipleSeqAlignment(biopython)"""
return MultipleSeqAlignment(
self.to_records(gap=True), annotations=self.meta_to_json()
)
to_bam(self, fname: str, ref_allele: str = '', save_ref: bool = True) -> None
inherited
Save the MSA into bam
All alleles will seen as reads aligned on ref_allele
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
fname |
str |
The name of bamfile |
required |
ref_allele |
str |
The reference allele. If the ref_allele is empty, the first allele will be reference. |
'' |
save_ref |
bool |
The reference allele will also be saved in the bam file |
True |
Source code in pyhlamsa/gene/genemsa.py
def to_bam(self, fname: str, ref_allele: str = "", save_ref: bool = True) -> None:
"""
Save the MSA into bam
All alleles will seen as reads aligned on `ref_allele`
Args:
fname (str): The name of bamfile
ref_allele (str): The reference allele.
If the ref_allele is empty, the first allele will be reference.
save_ref (bool): The reference allele will also be saved in the bam file
"""
ref_allele, ref_seq = self.get_allele_or_error(ref_allele)
# setup reference and header
header = {
"HD": {"VN": "1.0"},
"SQ": [
{"LN": len(ref_seq.replace("-", "").replace("E", "")), "SN": ref_allele}
],
}
# write bam file
with pysam.AlignmentFile(fname, "wb", header=header) as outf:
for allele, seq in self.items():
# skip
if not save_ref and allele == ref_allele:
continue
# init bam record
a = pysam.AlignedSegment()
a.query_name = allele
a.query_sequence = seq.replace("E", "").replace("-", "")
a.cigar = cigar.cigar_to_pysam(self.get_cigar(allele, ref_allele)) # type: ignore
# set to default
a.mapping_quality = 60
a.reference_id = 0
a.reference_start = 0
# a.template_length = 0
# a.query_qualities = [30] * len(a.query_sequence)
# a.flag = 0
# a.next_reference_id = 0
# a.next_reference_start = 0
outf.write(a)
pysam.sort("-o", fname, fname) # type: ignore
pysam.index(fname) # type: ignore
to_fasta(self, fname: FileType, gap: bool = True, ref_only: bool = False) -> None
inherited
Save the MSA into fasta
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
gap |
bool |
The sequence included gap or not |
True |
ref_only |
bool |
Save reference sequence only |
False |
Source code in pyhlamsa/gene/genemsa.py
def to_fasta(
self, fname: FileType, gap: bool = True, ref_only: bool = False
) -> None:
"""
Save the MSA into fasta
Args:
gap (bool): The sequence included gap or not
ref_only (bool): Save reference sequence only
"""
if ref_only:
msa = self.select_allele([self.get_reference()[0]])
else:
msa = self
SeqIO.write(msa.to_records(gap=gap), fname, "fasta")
to_gff(self, fname: FileType, strand: str = '+', ref_allele: str = '', igv_show_label: bool = False, save_all: bool = False) -> None
inherited
Save to GFF3 format
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
fname |
str |
The file name of gff3 |
required |
strand |
str |
Must be "+" or "-". If the strand is "-", it will add |
'+' |
ref_allele |
str |
The name of allele (Must be the same in save_bam) |
'' |
igv_show_label |
bool |
If it's false, it will generate proper GFF3. Set it for True as default for easiler label reading in IGV. |
False |
save_all |
bool |
Set it True if you want to create gff records for all alleles in msa Note that this is not very fast. |
False |
Source code in pyhlamsa/gene/genemsa.py
def to_gff(
self,
fname: FileType,
strand: str = "+",
ref_allele: str = "",
igv_show_label: bool = False,
save_all: bool = False,
) -> None:
"""
Save to GFF3 format
Args:
fname (str): The file name of gff3
strand (str): Must be "+" or "-".
If the strand is "-", it will add `strand` in GFF file,
if you want to reverse the sequence, please use `reverse_complement` first.
ref_allele (str): The name of allele (Must be the same in save_bam)
igv_show_label (bool): If it's false, it will generate proper GFF3.
Set it for True as default for easiler label reading in IGV.
save_all (bool):
Set it True if you want to create gff records for all alleles in msa
Note that this is not very fast.
"""
# TODO: should I save strand == '-' in model?
ref_allele = self.get_allele_or_error(ref_allele)[0]
# labels
if not all(b.type for b in self.blocks):
self.logger.warning(
"You should assign block's label. (We assume seq_type='other')"
)
self.assume_label("other")
if save_all:
alleles = list(self.list_alleles())
else:
alleles = [ref_allele]
records = []
for allele in alleles:
self.logger.debug(f"{allele} to gff")
# remove gap
msa = self.select_allele([allele]).shrink()
records.extend(
_block_to_gff(
msa.blocks, allele, strand=strand, igv_show_label=igv_show_label
)
)
# save
f_gff, require_close = getFileHandle(fname)
f_gff.write("##gff-version 3\n")
f_gff.write(_table_to_string(records))
if require_close:
f_gff.close()
to_imgt_alignment(self, file: FileType, seq_type: str = 'gen', index_start_from: str = 'exon1') -> None
inherited
Export to IMGT-alignment-like format.
But some features do not implemented: * AA codon position: So the values in the header always 1 * position: So the values in the header always 1
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
seq_type |
str |
gen or nuc format |
'gen' |
index_start_from |
str |
set the start position (1) starts from. |
'exon1' |
Source code in pyhlamsa/gene/genemsa.py
def to_imgt_alignment(
self,
file: FileType,
seq_type: str = "gen",
index_start_from: str = "exon1",
) -> None:
"""
Export to IMGT-alignment-like format.
But some features do not implemented:
* AA codon position: So the values in the header always 1
* position: So the values in the header always 1
Args:
seq_type: gen or nuc format
index_start_from: set the start position (1) starts from.
"""
# This function should be located in io_operation
new_msa = self._calc_imgt_alignment_msa()
start = new_msa.get_block_interval(index_start_from)[0]
# The position is calculated by reference sequence not MSA itself
ref_no_gap_pos = -1
seq = new_msa.get_reference()[1]
for i, ind in enumerate(new_msa.index):
if seq[i] != ".":
ref_no_gap_pos += 1
ind.pos = ref_no_gap_pos
if seq_type == "gen":
# force modify the index
# IMGT ends with -1 and start with 1
for ind in new_msa.index:
ind.pos -= start - 1
if ind.pos < 0:
ind.pos -= 1
pages = self._format_page(
[new_msa.index],
header_text_align="left",
index_header=["gDNA"],
show_position_when_same_value=False,
)
elif seq_type == "nuc":
fake_index = [IndexInfo(pos=0)] * len(new_msa.index)
pages = self._format_page(
[new_msa.index, fake_index],
header_text_align="left",
index_header=["cDNA", "AA Codon"],
show_position_when_same_value=False,
)
else:
raise NotImplementedError(f"Not implement {seq_type=}")
# write
txt, require_close = getFileHandle(file)
txt.writelines(chain.from_iterable(map(new_msa._apply_page, pages)))
if require_close:
txt.close()
to_records(self, gap: bool = True) -> list[SeqRecord]
inherited
Transfer MSA to list of SeqRecord.
If the base is E
which indicate the bases are not existed for exon-only sequence,
those beses are treated as gap.
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
gap |
bool |
The sequence included gap or not |
True |
Source code in pyhlamsa/gene/genemsa.py
def to_records(self, gap: bool = True) -> list[SeqRecord]:
"""
Transfer MSA to list of SeqRecord.
If the base is `E`
which indicate the bases are not existed for exon-only sequence,
those beses are treated as gap.
Args:
gap (bool): The sequence included gap or not
"""
if gap:
return [
SeqRecord(Seq(seq.replace("E", "-")), id=allele, description="")
for allele, seq in self.items()
]
else:
return [
SeqRecord(
Seq(seq.replace("E", "").replace("-", "")),
id=allele,
description="",
)
for allele, seq in self.items()
]
to_vcf(self, file_vcf: str, ref_allele: str = '', save_ref: bool = True, plain_text: bool = False) -> None
inherited
Save Genemsa into vcf format
It will save msa into sorted and normalized vcf file (xxx.vcf.gz) along with vcf's index (xxx.vcf.gz.tbi) (You can still output plain vcf without any manipulation by set plain_text=True)
Note that vcf-format discard the per-base alignment especially when there are many snps/indels mixed together in that position. Also, after vcf is normalized, the msa may not be the same MSA alignment as before. (But sequences are still the same)
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
ref_allele |
str |
The name of reference allele. I recommend the reference allele contains no gaps. |
'' |
save_ref |
bool |
The reference allele will also be saved in the vcf file |
True |
plain_text |
bool |
Disable sort vcf and index vcf |
False |
Source code in pyhlamsa/gene/genemsa.py
def to_vcf(
self,
file_vcf: str,
ref_allele: str = "",
save_ref: bool = True,
plain_text: bool = False,
) -> None:
"""
Save Genemsa into vcf format
It will save msa into sorted and normalized vcf file (xxx.vcf.gz)
along with vcf's index (xxx.vcf.gz.tbi)
(You can still output plain vcf without any manipulation by set plain_text=True)
Note that vcf-format discard the per-base alignment especially
when there are many snps/indels mixed together in that position.
Also, after vcf is normalized, the msa may not be the same MSA alignment as before.
(But sequences are still the same)
Args:
ref_allele (str):
The name of reference allele.
I recommend the reference allele contains no gaps.
save_ref (bool): The reference allele will also be saved in the vcf file
plain_text (bool): Disable sort vcf and index vcf
"""
ref_allele, ref_seq = self.get_allele_or_error(ref_allele)
# extract all variants from all alleles
allele_variants = {}
for allele in self.list_alleles():
if not save_ref and allele == ref_allele:
continue
variants = vcf.extract_variants(ref_seq, self.get(allele))
for v in variants:
v.chrom = ref_allele
allele_variants[allele] = variants
# remove suffix
if file_vcf.endswith(".vcf.gz"):
basename = file_vcf[:-7]
elif file_vcf.endswith(".vcf"):
basename = file_vcf[:-4]
else:
basename = file_vcf
tmpname = f"{basename}.tmp"
# to vcf
with open(f"{tmpname}.vcf", "w") as f_vcf:
f_vcf.write(vcf.get_vcf_header(ref_allele, ref_seq))
f_vcf.write("\n")
f_vcf.write(_table_to_string(vcf.variants_to_table(allele_variants)))
if plain_text:
os.rename(f"{tmpname}.vcf", f"{basename}.vcf")
return
# sort, normalize and index
self.select_allele([ref_allele]).to_fasta(f"{tmpname}.fa", gap=False)
with open(f"{tmpname}.norm.vcf.gz", "wb") as f_vcf:
f_vcf.write(
bcftools.norm( # type: ignore
"-f", f"{tmpname}.fa", f"{tmpname}.vcf", "-O", "z"
)
)
with open(f"{basename}.vcf.gz", "wb") as f_vcf:
f_vcf.write(bcftools.sort(f"{tmpname}.norm.vcf.gz", "-O", "z")) # type: ignore
bcftools.index(f"{basename}.vcf.gz", "-t", "-f") # type: ignore
os.remove(f"{tmpname}.fa")
os.remove(f"{tmpname}.fa.fai")
os.remove(f"{tmpname}.vcf")
os.remove(f"{tmpname}.norm.vcf.gz")
truth(self) -> bool
inherited
If msa has 0 alleles return False else return Implement if(self) function
Source code in pyhlamsa/gene/genemsa.py
def truth(self) -> bool:
"""
If msa has 0 alleles return False
else return Implement if(self) function
"""
return bool(self.alleles)
HLAmsa (Familymsa)
A HLA interface.
This module read the HLA MSA from alignments/*.txt files,
which use | to separate the intron/exon/UTR regions, but
the labels are assumed with the order:
5UTR exon1 intron1 exon2 ... exonN 3UTR
Attributes:
| Name | Type | Description |
|---|---|---|
genes |
dict[str, Genemsa] |
The dictionary use gene_name as key and msa object as value |
Source code in pyhlamsa/gene_family/hla.py
class HLAmsa(Familymsa):
"""
A HLA interface.
This module read the HLA MSA from alignments/*.txt files,
which use `|` to separate the intron/exon/UTR regions, but
the labels are assumed with the order:
`5UTR exon1 intron1 exon2 ... exonN 3UTR`
Attributes:
genes (dict[str, Genemsa]):
The dictionary use gene_name as key and msa object as value
"""
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = ["gen", "nuc"],
imgt_alignment_folder: str = "",
version: str = "Latest",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype (str | list[str] | set[str]): A list of filetype.
If both `gen` and `nuc` are given, it will merge them automatically.
imgt_alignment_folder (str): Path to your IMGT-formatted MSA alignment file
You can manually download the folder from
<https://github.com/ANHIG/IMGTHLA/tree/Latest/alignments>
or <http://ftp.ebi.ac.uk/pub/databases/ipd/imgt/hla/>
and unzip Alignments_Rel_3470.zip
Otherwise it will automatically download the database to
`imgt_alignment_folder`. Default is `./alignment_v{verion}`
version (str): IMGT version you want to download (e.g. 3470 for 3.47.0)
If `imgt_alignment_folder` is existed, this value will be ignored.
Use `Latest` to get latest version.
"""
if not imgt_alignment_folder:
imgt_alignment_folder = f"alignment_v{version}"
super().__init__(
genes, filetype, db_folder=imgt_alignment_folder, version=version
)
def _download_db(self, version: str = "Latest") -> None:
"""
Download the IMGTHLA alignments folder to `db_folder`
I get the alignments folder from git instead of
<http://ftp.ebi.ac.uk/pub/databases/ipd/imgt/hla/>
for better version controlling
"""
with TemporaryDirectory() as tmp_dir:
self._run_shell(
"git",
"clone",
"--branch",
version,
"--single-branch",
"https://github.com/ANHIG/IMGTHLA.git",
tmp_dir,
)
self._run_shell("mv", tmp_dir + "/alignments", self.db_folder)
def _get_name(self, search_name: str) -> set[str]:
"""Handy function to list names from file pattern"""
arr_files = glob(search_name)
return set([f.split("/")[-1].split("_")[0] for f in arr_files])
def list_db_gene(self, filetype: TypeSet) -> list[str]:
"""List the gene in folder"""
drb = set(["DRB1", "DRB3", "DRB4", "DRB5"])
if "gen" in filetype:
names = names_gen = self._get_name(f"{self.db_folder}/*_gen.txt") | drb
if "nuc" in filetype:
names = names_nuc = self._get_name(f"{self.db_folder}/*_nuc.txt") | drb
if "gen" in filetype and "nuc" in filetype:
names = names_gen & names_nuc
# * E exon7 is ambiguous, exon8 is gone (Also exon8 is not pseudo exon)
# <E gen alleles=258 block=5UTR(301) exon1(64) intron1(130)
# exon2(270) intron2(244) exon3(276) intron3(621) exon4(276)
# intron4(124) exon5(117) intron5(751) exon6(33) intron6(104)
# exon7(43) intron7(165) exon8(5) 3UTR(356)>
# <E nuc alleles=262 block=exon1(64) exon2(270) exon3(276)
# exon4(276) exon5(117) exon6(33) exon7(41)>
if "gen" in filetype and "nuc" in filetype:
names = names - set(["E"])
return list(sorted(names))
def read_db_gene(self, gene: str, filetype: TypeSet) -> Genemsa:
"""
Read `{gene}_{filetype}.txt`.
If both `gen` and `nuc` are given, it will merge them.
"""
if "gen" in filetype:
msa_gen = Genemsa.read_imgt_alignment(f"{self.db_folder}/{gene}_gen.txt")
msa_gen.gene_name = gene
if gene != "P":
# P: special case: has even block
# <P gen alleles=5 block=(475) (261) (589) (276) (124) (117)
# (412) (33) (150) (48) (163) (297)>
msa_gen.assume_label("gen")
self.logger.debug(f"Gen {msa_gen}")
if "nuc" in filetype:
# Special Case: DRB* nuc are in DRB_nuc.txt
if gene.startswith("DRB"):
msa_nuc = Genemsa.read_imgt_alignment(f"{self.db_folder}/DRB_nuc.txt")
msa_nuc = msa_nuc.select_allele(gene + ".*")
else:
msa_nuc = Genemsa.read_imgt_alignment(
f"{self.db_folder}/{gene}_nuc.txt"
)
msa_nuc.gene_name = gene
msa_nuc.assume_label("nuc")
self.logger.debug(f"Nuc {msa_nuc}")
if "gen" in filetype and "nuc" in filetype:
# remove some gene
diff_name = list(
set(msa_gen.get_sequence_names()) - set(msa_nuc.get_sequence_names())
)
if diff_name:
self.logger.warning(
f"Remove alleles doesn't exist in gen and nuc either: {diff_name}"
)
msa_gen = msa_gen.remove_allele(diff_name)
# merge
msa_merged = msa_gen.merge_exon(msa_nuc)
self.logger.debug(f"Merged {msa_merged}")
return msa_merged
elif "gen" in filetype:
return msa_gen
elif "nuc" in filetype:
return msa_nuc
raise ValueError("gen or nuc are not exist in filetype")
__getitem__(self, index: str) -> Genemsa
inherited
special
Source code in pyhlamsa/gene_family/hla.py
def __getitem__(self, index: str) -> Genemsa:
"""Get specific gene's msa"""
return self.genes[index]
__init__(self, genes: Union[str, Iterable[str]] = None, filetype: Union[str, Iterable[str]] = ['gen', 'nuc'], imgt_alignment_folder: str = '', version: str = 'Latest')
special
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
genes |
str | list[str] |
A list of genes you want to read. Set None if you want read all gene in HLA |
None |
filetype |
str | list[str] | set[str] |
A list of filetype. If both |
['gen', 'nuc'] |
imgt_alignment_folder |
str |
Path to your IMGT-formatted MSA alignment file You can manually download the folder from https://github.com/ANHIG/IMGTHLA/tree/Latest/alignments or http://ftp.ebi.ac.uk/pub/databases/ipd/imgt/hla/ and unzip Alignments_Rel_3470.zip Otherwise it will automatically download the database to
|
'' |
version |
str |
IMGT version you want to download (e.g. 3470 for 3.47.0) If |
'Latest' |
Source code in pyhlamsa/gene_family/hla.py
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = ["gen", "nuc"],
imgt_alignment_folder: str = "",
version: str = "Latest",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype (str | list[str] | set[str]): A list of filetype.
If both `gen` and `nuc` are given, it will merge them automatically.
imgt_alignment_folder (str): Path to your IMGT-formatted MSA alignment file
You can manually download the folder from
<https://github.com/ANHIG/IMGTHLA/tree/Latest/alignments>
or <http://ftp.ebi.ac.uk/pub/databases/ipd/imgt/hla/>
and unzip Alignments_Rel_3470.zip
Otherwise it will automatically download the database to
`imgt_alignment_folder`. Default is `./alignment_v{verion}`
version (str): IMGT version you want to download (e.g. 3470 for 3.47.0)
If `imgt_alignment_folder` is existed, this value will be ignored.
Use `Latest` to get latest version.
"""
if not imgt_alignment_folder:
imgt_alignment_folder = f"alignment_v{version}"
super().__init__(
genes, filetype, db_folder=imgt_alignment_folder, version=version
)
__iter__(self) -> Iterable[str]
inherited
special
Source code in pyhlamsa/gene_family/hla.py
def __iter__(self) -> Iterable[str]:
"""Iter gene name like iter(dict)"""
return iter(self.genes)
items(self) -> Iterable[tuple[str, pyhlamsa.gene.genemsa.Genemsa]]
inherited
list gene name and msa like dict.items()
Source code in pyhlamsa/gene_family/hla.py
def items(self) -> Iterable[tuple[str, Genemsa]]:
"""list gene name and msa like dict.items()"""
return self.genes.items()
list_db_gene(self, filetype: Union[str, Iterable[str]]) -> list
List the gene in folder
Source code in pyhlamsa/gene_family/hla.py
def list_db_gene(self, filetype: TypeSet) -> list[str]:
"""List the gene in folder"""
drb = set(["DRB1", "DRB3", "DRB4", "DRB5"])
if "gen" in filetype:
names = names_gen = self._get_name(f"{self.db_folder}/*_gen.txt") | drb
if "nuc" in filetype:
names = names_nuc = self._get_name(f"{self.db_folder}/*_nuc.txt") | drb
if "gen" in filetype and "nuc" in filetype:
names = names_gen & names_nuc
# * E exon7 is ambiguous, exon8 is gone (Also exon8 is not pseudo exon)
# <E gen alleles=258 block=5UTR(301) exon1(64) intron1(130)
# exon2(270) intron2(244) exon3(276) intron3(621) exon4(276)
# intron4(124) exon5(117) intron5(751) exon6(33) intron6(104)
# exon7(43) intron7(165) exon8(5) 3UTR(356)>
# <E nuc alleles=262 block=exon1(64) exon2(270) exon3(276)
# exon4(276) exon5(117) exon6(33) exon7(41)>
if "gen" in filetype and "nuc" in filetype:
names = names - set(["E"])
return list(sorted(names))
list_genes(self) -> list
inherited
List all the gene's name in this family
Source code in pyhlamsa/gene_family/hla.py
def list_genes(self) -> list[str]:
"""List all the gene's name in this family"""
return list(self.genes.keys())
read_db_gene(self, gene: str, filetype: Union[str, Iterable[str]]) -> Genemsa
Read {gene}_{filetype}.txt.
If both gen and nuc are given, it will merge them.
Source code in pyhlamsa/gene_family/hla.py
def read_db_gene(self, gene: str, filetype: TypeSet) -> Genemsa:
"""
Read `{gene}_{filetype}.txt`.
If both `gen` and `nuc` are given, it will merge them.
"""
if "gen" in filetype:
msa_gen = Genemsa.read_imgt_alignment(f"{self.db_folder}/{gene}_gen.txt")
msa_gen.gene_name = gene
if gene != "P":
# P: special case: has even block
# <P gen alleles=5 block=(475) (261) (589) (276) (124) (117)
# (412) (33) (150) (48) (163) (297)>
msa_gen.assume_label("gen")
self.logger.debug(f"Gen {msa_gen}")
if "nuc" in filetype:
# Special Case: DRB* nuc are in DRB_nuc.txt
if gene.startswith("DRB"):
msa_nuc = Genemsa.read_imgt_alignment(f"{self.db_folder}/DRB_nuc.txt")
msa_nuc = msa_nuc.select_allele(gene + ".*")
else:
msa_nuc = Genemsa.read_imgt_alignment(
f"{self.db_folder}/{gene}_nuc.txt"
)
msa_nuc.gene_name = gene
msa_nuc.assume_label("nuc")
self.logger.debug(f"Nuc {msa_nuc}")
if "gen" in filetype and "nuc" in filetype:
# remove some gene
diff_name = list(
set(msa_gen.get_sequence_names()) - set(msa_nuc.get_sequence_names())
)
if diff_name:
self.logger.warning(
f"Remove alleles doesn't exist in gen and nuc either: {diff_name}"
)
msa_gen = msa_gen.remove_allele(diff_name)
# merge
msa_merged = msa_gen.merge_exon(msa_nuc)
self.logger.debug(f"Merged {msa_merged}")
return msa_merged
elif "gen" in filetype:
return msa_gen
elif "nuc" in filetype:
return msa_nuc
raise ValueError("gen or nuc are not exist in filetype")
HLAmsaEX (Familymsa)
A HLA interface but read gene from MSF and read intron/exon information from hla.dat
I think this one is more reliable
Attributes:
| Name | Type | Description |
|---|---|---|
genes |
dict[str, Genemsa] |
The dictionary use gene_name as key and msa object as value |
Source code in pyhlamsa/gene_family/hla_ex.py
class HLAmsaEX(Familymsa):
"""
A HLA interface but read gene from MSF and
read intron/exon information from hla.dat
I think this one is more reliable
Attributes:
genes (dict[str, Genemsa]):
The dictionary use gene_name as key and msa object as value
"""
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = ["gen", "nuc"],
imgt_folder: str = "",
version: str = "Latest",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype (str | list[str] | set[str]): A list of filetype.
If both `gen` and `nuc` are given, it will merge them automatically.
imgt_folder (str): Path to your IMGT/HLA root folder
You can manually download <https://github.com/ANHIG/IMGTHLA/> and
checkout to specific branch
Or it will automatically download the database with assigned `version` to
`imgt_folder`. Default is `./IMGT_v{verion}`
version (str): IMGT version you want to download (e.g. 3470 for 3.47.0)
If `imgt_folder` is existed, this value will be ignored.
You can use `Latest` to get the latest version
"""
if not imgt_folder:
imgt_folder = f"IMGT_v{version}"
super().__init__(genes, filetype, db_folder=imgt_folder, version=version)
def _download_db(self, version: str = "Latest") -> None:
"""
Download the IMGT/HLA msf and hla.dat to folder `imgt_folder`
"""
try:
self._run_shell("git", "lfs")
except subprocess.CalledProcessError:
self.logger.error(f"git lfs not installed (Try apt install git-lfs)")
exit()
self._run_shell(
"git",
"clone",
"--branch",
version,
"--single-branch",
"https://github.com/ANHIG/IMGTHLA.git",
self.db_folder,
)
self._run_shell("git", "lfs", "pull", cwd=self.db_folder)
def _get_name(self, search_name: str) -> set[str]:
"""Extract name from file pattern"""
arr_files = glob(search_name)
return set([f.split("/")[-1].split("_")[0] for f in arr_files])
def list_db_gene(self, filetype: TypeSet) -> list[str]:
"""List the gene in folder"""
drb = set(["DRB1", "DRB3", "DRB4", "DRB5"])
if "gen" in filetype:
names = names_gen = self._get_name(f"{self.db_folder}/msf/*_gen.msf") | drb
if "nuc" in filetype:
names = names_nuc = self._get_name(f"{self.db_folder}/msf/*_nuc.msf") | drb
if "gen" in filetype and "nuc" in filetype:
# HLA-E nuc has 7 bases differences from exon7 and exon8
names = (names_gen & names_nuc) - set("E")
return sorted(names)
def read_db_gene(self, gene: str, filetype: TypeSet) -> Genemsa:
"""
Read `msf/{gene}_{filetype}.msf` and hla.dat.
If both `gen` and `nuc` are given, it will merge them.
"""
if not hasattr(self, "dat"):
self.logger.debug(f"Reading hla.dat")
self.dat = dat.read_dat_block(f"{self.db_folder}/hla.dat")
if "gen" in filetype:
msa_gen = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_gen.msf")
msa_gen.gene_name = gene
msa_gen = dat.apply_dat_info_on_msa(msa_gen, self.dat, seq_type="gen")
self.logger.debug(f"{msa_gen}")
if "nuc" in filetype:
# Special Case: DRB* nuc are in DRB_nuc.txt
if gene.startswith("DRB"):
msa_nuc = Genemsa.read_msf_file(f"{self.db_folder}/msf/DRB_nuc.msf")
msa_nuc = msa_nuc.select_allele(gene + ".*")
else:
msa_nuc = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_nuc.msf")
msa_nuc.gene_name = gene
msa_nuc = dat.apply_dat_info_on_msa(msa_nuc, self.dat, seq_type="nuc")
self.logger.debug(f"{msa_nuc}")
if "gen" in filetype and "nuc" in filetype:
# remove some gen not included in nuc
diff_name = list(
set(msa_gen.get_sequence_names()) - set(msa_nuc.get_sequence_names())
)
if diff_name:
self.logger.warning(
f"Remove alleles existed in gen but not in nuc: {diff_name}"
)
msa_gen = msa_gen.remove_allele(diff_name)
# merge
msa_merged = msa_gen.merge_exon(msa_nuc)
self.logger.debug(f"{msa_merged}")
return msa_merged
elif "gen" in filetype:
return msa_gen
elif "nuc" in filetype:
return msa_nuc
raise ValueError("gen or nuc are not exist in filetype")
__getitem__(self, index: str) -> Genemsa
inherited
special
Source code in pyhlamsa/gene_family/hla_ex.py
def __getitem__(self, index: str) -> Genemsa:
"""Get specific gene's msa"""
return self.genes[index]
__init__(self, genes: Union[str, Iterable[str]] = None, filetype: Union[str, Iterable[str]] = ['gen', 'nuc'], imgt_folder: str = '', version: str = 'Latest')
special
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
genes |
str | list[str] |
A list of genes you want to read. Set None if you want read all gene in HLA |
None |
filetype |
str | list[str] | set[str] |
A list of filetype. If both |
['gen', 'nuc'] |
imgt_folder |
str |
Path to your IMGT/HLA root folder You can manually download https://github.com/ANHIG/IMGTHLA/ and checkout to specific branch Or it will automatically download the database with assigned |
'' |
version |
str |
IMGT version you want to download (e.g. 3470 for 3.47.0) If You can use |
'Latest' |
Source code in pyhlamsa/gene_family/hla_ex.py
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = ["gen", "nuc"],
imgt_folder: str = "",
version: str = "Latest",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype (str | list[str] | set[str]): A list of filetype.
If both `gen` and `nuc` are given, it will merge them automatically.
imgt_folder (str): Path to your IMGT/HLA root folder
You can manually download <https://github.com/ANHIG/IMGTHLA/> and
checkout to specific branch
Or it will automatically download the database with assigned `version` to
`imgt_folder`. Default is `./IMGT_v{verion}`
version (str): IMGT version you want to download (e.g. 3470 for 3.47.0)
If `imgt_folder` is existed, this value will be ignored.
You can use `Latest` to get the latest version
"""
if not imgt_folder:
imgt_folder = f"IMGT_v{version}"
super().__init__(genes, filetype, db_folder=imgt_folder, version=version)
__iter__(self) -> Iterable[str]
inherited
special
Source code in pyhlamsa/gene_family/hla_ex.py
def __iter__(self) -> Iterable[str]:
"""Iter gene name like iter(dict)"""
return iter(self.genes)
items(self) -> Iterable[tuple[str, pyhlamsa.gene.genemsa.Genemsa]]
inherited
list gene name and msa like dict.items()
Source code in pyhlamsa/gene_family/hla_ex.py
def items(self) -> Iterable[tuple[str, Genemsa]]:
"""list gene name and msa like dict.items()"""
return self.genes.items()
list_db_gene(self, filetype: Union[str, Iterable[str]]) -> list
List the gene in folder
Source code in pyhlamsa/gene_family/hla_ex.py
def list_db_gene(self, filetype: TypeSet) -> list[str]:
"""List the gene in folder"""
drb = set(["DRB1", "DRB3", "DRB4", "DRB5"])
if "gen" in filetype:
names = names_gen = self._get_name(f"{self.db_folder}/msf/*_gen.msf") | drb
if "nuc" in filetype:
names = names_nuc = self._get_name(f"{self.db_folder}/msf/*_nuc.msf") | drb
if "gen" in filetype and "nuc" in filetype:
# HLA-E nuc has 7 bases differences from exon7 and exon8
names = (names_gen & names_nuc) - set("E")
return sorted(names)
list_genes(self) -> list
inherited
List all the gene's name in this family
Source code in pyhlamsa/gene_family/hla_ex.py
def list_genes(self) -> list[str]:
"""List all the gene's name in this family"""
return list(self.genes.keys())
read_db_gene(self, gene: str, filetype: Union[str, Iterable[str]]) -> Genemsa
Read msf/{gene}_{filetype}.msf and hla.dat.
If both gen and nuc are given, it will merge them.
Source code in pyhlamsa/gene_family/hla_ex.py
def read_db_gene(self, gene: str, filetype: TypeSet) -> Genemsa:
"""
Read `msf/{gene}_{filetype}.msf` and hla.dat.
If both `gen` and `nuc` are given, it will merge them.
"""
if not hasattr(self, "dat"):
self.logger.debug(f"Reading hla.dat")
self.dat = dat.read_dat_block(f"{self.db_folder}/hla.dat")
if "gen" in filetype:
msa_gen = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_gen.msf")
msa_gen.gene_name = gene
msa_gen = dat.apply_dat_info_on_msa(msa_gen, self.dat, seq_type="gen")
self.logger.debug(f"{msa_gen}")
if "nuc" in filetype:
# Special Case: DRB* nuc are in DRB_nuc.txt
if gene.startswith("DRB"):
msa_nuc = Genemsa.read_msf_file(f"{self.db_folder}/msf/DRB_nuc.msf")
msa_nuc = msa_nuc.select_allele(gene + ".*")
else:
msa_nuc = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_nuc.msf")
msa_nuc.gene_name = gene
msa_nuc = dat.apply_dat_info_on_msa(msa_nuc, self.dat, seq_type="nuc")
self.logger.debug(f"{msa_nuc}")
if "gen" in filetype and "nuc" in filetype:
# remove some gen not included in nuc
diff_name = list(
set(msa_gen.get_sequence_names()) - set(msa_nuc.get_sequence_names())
)
if diff_name:
self.logger.warning(
f"Remove alleles existed in gen but not in nuc: {diff_name}"
)
msa_gen = msa_gen.remove_allele(diff_name)
# merge
msa_merged = msa_gen.merge_exon(msa_nuc)
self.logger.debug(f"{msa_merged}")
return msa_merged
elif "gen" in filetype:
return msa_gen
elif "nuc" in filetype:
return msa_nuc
raise ValueError("gen or nuc are not exist in filetype")
KIRmsa (Familymsa)
A KIR interface that read MSA from MSF and KIR.dat
Attributes:
| Name | Type | Description |
|---|---|---|
genes |
dict[str, Genemsa] |
The msa object for each gene |
Source code in pyhlamsa/gene_family/kir.py
class KIRmsa(Familymsa):
"""
A KIR interface that read MSA from MSF and KIR.dat
Attributes:
genes (dict[str, Genemsa]): The msa object for each gene
"""
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = ["gen", "nuc"],
ipd_folder: str = "",
version: str = "Latest",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype (str | list[str] | set[str]): A list of filetype.
If both `gen` and `nuc` are given, it will merge them automatically.
ipd_folder (str): Path to your IPD/KIR folder
You can manually download <https://github.com/ANHIG/IPDKIR> and
checkout to specific branch
Or it will automatically download the database with assigned `version` to
`ipd_folder`. Default is `./kIR_v{verion}`
version (str): IMGT version you want to download (2110 for version 2.11.0)
If `ipd_folder` is existed, this value will be ignored.
version='Latest' to get latest version, however sometime it cannot work
because database may change the format, or contains bugs
(e.g. 2.11.0 https://github.com/ANHIG/IPDKIR/issues/44)
"""
# Why not version 2110 -> 2DL4,2DL5 has exon 4
if not ipd_folder:
ipd_folder = f"KIR_v{version}"
super().__init__(genes, filetype, db_folder=ipd_folder, version=version)
def _download_db(self, version: str = "Latest") -> None:
"""
Download the KIR to `IPDKIR`
"""
self._run_shell(
"git",
"clone",
"--branch",
version,
"--single-branch",
"https://github.com/ANHIG/IPDKIR",
self.db_folder,
)
def _get_name(self, search_name: str) -> set[str]:
"""Extract name from file pattern"""
arr_files = glob(search_name)
return set([f.split("/")[-1].split("_")[0] for f in arr_files])
def list_db_gene(self, filetype: TypeSet) -> list[str]:
"""List the gene in folder"""
if "gen" in filetype:
names = names_gen = self._get_name(f"{self.db_folder}/msf/*_gen.msf")
if "nuc" in filetype:
# Most's of E nuc is different from dat record
names = names_nuc = self._get_name(f"{self.db_folder}/msf/*_nuc.msf")
if "gen" in filetype and "nuc" in filetype:
names = names_gen & names_nuc
return sorted(names)
def read_db_gene(self, gene: str, filetype: TypeSet) -> Genemsa:
"""
Read `{gene}_{filetype}.msf and kir.dat`.
If both `gen` and `nuc` are given, it will merge them.
"""
if not hasattr(self, "dat"):
self.logger.debug(f"Reading kir.dat")
if os.path.exists(f"{self.db_folder}/KIR.dat"):
self.dat = dat.read_dat_block(f"{self.db_folder}/KIR.dat")
else:
self.dat = dat.read_dat_block(f"{self.db_folder}/kir.dat")
if "gen" in filetype:
msa_gen = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_gen.msf")
msa_gen.gene_name = gene
msa_gen = dat.apply_dat_info_on_msa(msa_gen, self.dat, seq_type="gen")
self.logger.debug(f"Gen {msa_gen}")
if "nuc" in filetype:
msa_nuc = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_nuc.msf")
msa_nuc.gene_name = gene
msa_nuc = dat.apply_dat_info_on_msa(msa_nuc, self.dat, seq_type="nuc")
self.logger.debug(f"Nuc {msa_nuc}")
if "gen" in filetype and "nuc" in filetype:
# remove some gen not included in nuc
diff_name = list(
set(msa_gen.get_sequence_names()) - set(msa_nuc.get_sequence_names())
)
if diff_name:
self.logger.warning(
f"Remove alleles doesn't exist in gen and nuc either: {diff_name}"
)
msa_gen = msa_gen.remove_allele(diff_name)
# specical case
# exon 3 is pseudo exon
# so, fill with gene's exon3
gene_has_pseudo_exon3 = [
"KIR2DL1",
"KIR2DL2",
"KIR2DL3",
"KIR2DP1",
"KIR2DS1",
"KIR2DS2",
"KIR2DS3",
"KIR2DS4",
"KIR2DS5",
]
if gene in gene_has_pseudo_exon3:
exon3 = msa_gen.select_block([5])
for name in set(msa_nuc) - set(msa_gen):
exon3.append(name, "-" * exon3.get_length())
msa_nuc = (
msa_nuc.select_block(list(range(0, 2)))
+ exon3
+ msa_nuc.select_block(list(range(3, len(msa_nuc.blocks))))
)
# merge
msa_merged = msa_gen.merge_exon(msa_nuc)
self.logger.debug(f"Merged {msa_merged}")
return msa_merged
elif "gen" in filetype:
return msa_gen
elif "nuc" in filetype:
return msa_nuc
raise ValueError("gen or nuc are not exist in filetype")
__getitem__(self, index: str) -> Genemsa
inherited
special
Source code in pyhlamsa/gene_family/kir.py
def __getitem__(self, index: str) -> Genemsa:
"""Get specific gene's msa"""
return self.genes[index]
__init__(self, genes: Union[str, Iterable[str]] = None, filetype: Union[str, Iterable[str]] = ['gen', 'nuc'], ipd_folder: str = '', version: str = 'Latest')
special
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
genes |
str | list[str] |
A list of genes you want to read. Set None if you want read all gene in HLA |
None |
filetype |
str | list[str] | set[str] |
A list of filetype. If both |
['gen', 'nuc'] |
ipd_folder |
str |
Path to your IPD/KIR folder You can manually download https://github.com/ANHIG/IPDKIR and checkout to specific branch Or it will automatically download the database with assigned |
'' |
version |
str |
IMGT version you want to download (2110 for version 2.11.0) If version='Latest' to get latest version, however sometime it cannot work because database may change the format, or contains bugs (e.g. 2.11.0 https://github.com/ANHIG/IPDKIR/issues/44) |
'Latest' |
Source code in pyhlamsa/gene_family/kir.py
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = ["gen", "nuc"],
ipd_folder: str = "",
version: str = "Latest",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype (str | list[str] | set[str]): A list of filetype.
If both `gen` and `nuc` are given, it will merge them automatically.
ipd_folder (str): Path to your IPD/KIR folder
You can manually download <https://github.com/ANHIG/IPDKIR> and
checkout to specific branch
Or it will automatically download the database with assigned `version` to
`ipd_folder`. Default is `./kIR_v{verion}`
version (str): IMGT version you want to download (2110 for version 2.11.0)
If `ipd_folder` is existed, this value will be ignored.
version='Latest' to get latest version, however sometime it cannot work
because database may change the format, or contains bugs
(e.g. 2.11.0 https://github.com/ANHIG/IPDKIR/issues/44)
"""
# Why not version 2110 -> 2DL4,2DL5 has exon 4
if not ipd_folder:
ipd_folder = f"KIR_v{version}"
super().__init__(genes, filetype, db_folder=ipd_folder, version=version)
__iter__(self) -> Iterable[str]
inherited
special
Source code in pyhlamsa/gene_family/kir.py
def __iter__(self) -> Iterable[str]:
"""Iter gene name like iter(dict)"""
return iter(self.genes)
items(self) -> Iterable[tuple[str, pyhlamsa.gene.genemsa.Genemsa]]
inherited
list gene name and msa like dict.items()
Source code in pyhlamsa/gene_family/kir.py
def items(self) -> Iterable[tuple[str, Genemsa]]:
"""list gene name and msa like dict.items()"""
return self.genes.items()
list_db_gene(self, filetype: Union[str, Iterable[str]]) -> list
List the gene in folder
Source code in pyhlamsa/gene_family/kir.py
def list_db_gene(self, filetype: TypeSet) -> list[str]:
"""List the gene in folder"""
if "gen" in filetype:
names = names_gen = self._get_name(f"{self.db_folder}/msf/*_gen.msf")
if "nuc" in filetype:
# Most's of E nuc is different from dat record
names = names_nuc = self._get_name(f"{self.db_folder}/msf/*_nuc.msf")
if "gen" in filetype and "nuc" in filetype:
names = names_gen & names_nuc
return sorted(names)
list_genes(self) -> list
inherited
List all the gene's name in this family
Source code in pyhlamsa/gene_family/kir.py
def list_genes(self) -> list[str]:
"""List all the gene's name in this family"""
return list(self.genes.keys())
read_db_gene(self, gene: str, filetype: Union[str, Iterable[str]]) -> Genemsa
Read {gene}_{filetype}.msf and kir.dat.
If both gen and nuc are given, it will merge them.
Source code in pyhlamsa/gene_family/kir.py
def read_db_gene(self, gene: str, filetype: TypeSet) -> Genemsa:
"""
Read `{gene}_{filetype}.msf and kir.dat`.
If both `gen` and `nuc` are given, it will merge them.
"""
if not hasattr(self, "dat"):
self.logger.debug(f"Reading kir.dat")
if os.path.exists(f"{self.db_folder}/KIR.dat"):
self.dat = dat.read_dat_block(f"{self.db_folder}/KIR.dat")
else:
self.dat = dat.read_dat_block(f"{self.db_folder}/kir.dat")
if "gen" in filetype:
msa_gen = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_gen.msf")
msa_gen.gene_name = gene
msa_gen = dat.apply_dat_info_on_msa(msa_gen, self.dat, seq_type="gen")
self.logger.debug(f"Gen {msa_gen}")
if "nuc" in filetype:
msa_nuc = Genemsa.read_msf_file(f"{self.db_folder}/msf/{gene}_nuc.msf")
msa_nuc.gene_name = gene
msa_nuc = dat.apply_dat_info_on_msa(msa_nuc, self.dat, seq_type="nuc")
self.logger.debug(f"Nuc {msa_nuc}")
if "gen" in filetype and "nuc" in filetype:
# remove some gen not included in nuc
diff_name = list(
set(msa_gen.get_sequence_names()) - set(msa_nuc.get_sequence_names())
)
if diff_name:
self.logger.warning(
f"Remove alleles doesn't exist in gen and nuc either: {diff_name}"
)
msa_gen = msa_gen.remove_allele(diff_name)
# specical case
# exon 3 is pseudo exon
# so, fill with gene's exon3
gene_has_pseudo_exon3 = [
"KIR2DL1",
"KIR2DL2",
"KIR2DL3",
"KIR2DP1",
"KIR2DS1",
"KIR2DS2",
"KIR2DS3",
"KIR2DS4",
"KIR2DS5",
]
if gene in gene_has_pseudo_exon3:
exon3 = msa_gen.select_block([5])
for name in set(msa_nuc) - set(msa_gen):
exon3.append(name, "-" * exon3.get_length())
msa_nuc = (
msa_nuc.select_block(list(range(0, 2)))
+ exon3
+ msa_nuc.select_block(list(range(3, len(msa_nuc.blocks))))
)
# merge
msa_merged = msa_gen.merge_exon(msa_nuc)
self.logger.debug(f"Merged {msa_merged}")
return msa_merged
elif "gen" in filetype:
return msa_gen
elif "nuc" in filetype:
return msa_nuc
raise ValueError("gen or nuc are not exist in filetype")
CYPmsa (Familymsa)
A CYP interface that read MSA from fasta and haplotypes.tsv
Attributes:
| Name | Type | Description |
|---|---|---|
genes |
dict[str, Genemsa] |
The msa object for each gene |
Source code in pyhlamsa/gene_family/cyp.py
class CYPmsa(Familymsa):
"""
A CYP interface that read MSA from fasta and haplotypes.tsv
Attributes:
genes (dict[str, Genemsa]): The msa object for each gene
"""
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = [],
pharmvar_folder: str = "",
version: str = "5.2.2",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype: Ignore
pharmvar_folder (str): Path to your pharmvar folder
You should manually download <https://www.pharmvar.org/download>
and unzip it.
version (str): PharmVar version you want to download
Not works now
"""
if not pharmvar_folder:
pharmvar_folder = f"pharmvar-{version}"
super().__init__(genes, filetype, db_folder=pharmvar_folder, version=version)
def _download_db(self, version: str = "") -> None:
"""
Download the CYP from https://www.pharmvar.org/download
"""
raise ValueError(
"You should download CYP genes from https://www.pharmvar.org/download"
)
def list_db_gene(self, filetype: TypeSet = []) -> list[str]:
"""List the gene in folder"""
return sorted(os.listdir(self.db_folder))
def read_db_gene(self, gene: str, filetype: TypeSet = []) -> Genemsa:
"""
Read `{gene}/{gene}.haplotypes.fasta` and `{gene}/{gene}.haplotypes.tsv`
`filetype` will be ignored now
"""
# read fasta
ref_seqs = {}
for seq in SeqIO.parse(
f"{self.db_folder}/{gene}/{gene}.haplotypes.fasta", "fasta"
):
# split allele name
# e.g. rs75017182, rs56038477 PV01077 NG_008807.2 PharmVar Version:5.1.10
for name in seq.description.replace(", ", ",").split(" ")[0].split(","):
ref_seqs[name.strip()] = str(seq.seq)
self.logger.debug(f"Read sequence {ref_seqs.keys()}")
# read tsv
# split by '\t' and ignore header
var_text = open(glob(f"{self.db_folder}/{gene}/RefSeqGene/*.haplotypes.tsv")[0])
table = [i.strip().split("\t") for i in var_text if not i.startswith("#")][1:]
# Get Reference sequence
reference_seq = None
reference_name = None
for i in table:
if i[0] not in ref_seqs:
continue
if i[3] == "REFERENCE" and ref_seqs[i[0]][0]:
if reference_seq:
assert reference_seq == ref_seqs[i[0]]
continue
reference_name = i[0]
reference_seq = ref_seqs[reference_name]
if not reference_seq:
# hard-coded for DPYD, because it's reference `NG_008807.2` is missing
if gene == "DPYD":
reference_name = "rs111858276"
reference_seq = ref_seqs[reference_name]
reference_seq = (
reference_seq[: 376460 - 1] + "A" + reference_seq[376460:]
)
else:
raise ValueError(f"Not reference found in {gene}")
assert reference_name is not None
# Fill with Reference and reference is the first one
alleles = {reference_name: ""}
alleles.update(
{allele_name: reference_seq for allele_name in set(i[0] for i in table)}
)
self.logger.debug(f"Read vcf {alleles.keys()}")
# Remove duplciate
# in 5.1.10, there exist two same row
# CYP2D6*149 CYP2D6 rs59421388 NG_008376.4 8203 8203 G A substitution
# CYP2D6*149 CYP2D6 rs59421388 NG_008376.4 8203 8203 G A substitution
table_unique1 = filter(lambda i: i[3] != "REFERENCE", table)
table_unique2 = cast(tuple, map(tuple, table_unique1)) # type: ignore
table_unique = cast(list, map(list, set(list(table_unique2)))) # type: ignore
# Reconstruct msa from VCF variant
# VCF type: substitution and deleteion, insertion
# Table Header: ['Haplotype Name', 'Gene', 'rsID', 'ReferenceSequence',
# 'Variant Start', 'Variant Stop', 'Reference Allele', 'Variant Allele', 'Type']
# Insertion will move the index, so insert from last position first
for i in sorted(table_unique, key=lambda i: -int(i[4])):
if i[8] == "substitution":
pos = int(i[4]) - 1
end = pos + 1
elif i[8] == "deletion":
pos = int(i[4]) - 1
end = pos + len(i[6])
i[7] = "-" * len(i[6])
elif i[8] == "insertion":
# hard coded bugs
if i[0].startswith("CYP2A6*31"):
# CYP2A6*31.001 4530 4552 CCCCTTCCTGAGACCCTTAACCC - deletion
# CYP2A6*31.001 4530 4531 - AATCCATATGTGGAATCTG insertion
continue
if i[0].startswith("CYP2A6*27"):
# CYP2A6*27.001 7183 7184 - A insertion
continue
pos = int(i[4])
end = int(i[4]) + len(i[7])
i[6] = "-" * len(i[7])
# check the gap is already inserted by others
# if false: insert '-' for all alleles
# else: replace the '-' with insertion seq
if set(alleles[i[0]][pos:end]) != set("-"):
for allele_name in alleles:
alleles[allele_name] = (
alleles[allele_name][:pos]
+ i[6]
+ alleles[allele_name][pos:]
)
else:
raise ValueError(f"Unknown Type {i[8]}")
assert alleles[i[0]][pos:end] == i[6]
alleles[i[0]] = alleles[i[0]][:pos] + i[7] + alleles[i[0]][end:]
# split allele name (alleles.key() will change)
# e.g. rs75017182, rs56038477 DPYD rs75017182 NG_008807.2 346167
for allele_name in list(alleles.keys()):
if ", " in allele_name:
for an in allele_name.split(","):
alleles[an.strip()] = alleles[allele_name]
del alleles[allele_name]
# double check
length = None
for allele_name in alleles:
if length is None:
length = len(alleles[allele_name])
else:
assert length == len(alleles[allele_name])
if alleles[allele_name].replace("-", "") != ref_seqs[allele_name]:
raise ValueError(f"{allele_name} is not same as reference")
assert length
msa = Genemsa(gene)
msa.alleles = alleles
msa.blocks = [BlockInfo(length=length)]
return msa.assume_label("other")
__getitem__(self, index: str) -> Genemsa
inherited
special
Source code in pyhlamsa/gene_family/cyp.py
def __getitem__(self, index: str) -> Genemsa:
"""Get specific gene's msa"""
return self.genes[index]
__init__(self, genes: Union[str, Iterable[str]] = None, filetype: Union[str, Iterable[str]] = [], pharmvar_folder: str = '', version: str = '5.2.2')
special
Parameters:
| Name | Type | Description | Default |
|---|---|---|---|
genes |
str | list[str] |
A list of genes you want to read. Set None if you want read all gene in HLA |
None |
filetype |
Union[str, Iterable[str]] |
Ignore |
[] |
pharmvar_folder |
str |
Path to your pharmvar folder You should manually download https://www.pharmvar.org/download and unzip it. |
'' |
version |
str |
PharmVar version you want to download Not works now |
'5.2.2' |
Source code in pyhlamsa/gene_family/cyp.py
def __init__(
self,
genes: GeneSet = None,
filetype: TypeSet = [],
pharmvar_folder: str = "",
version: str = "5.2.2",
):
"""
Args:
genes (str | list[str]): A list of genes you want to read.
Set None if you want read all gene in HLA
filetype: Ignore
pharmvar_folder (str): Path to your pharmvar folder
You should manually download <https://www.pharmvar.org/download>
and unzip it.
version (str): PharmVar version you want to download
Not works now
"""
if not pharmvar_folder:
pharmvar_folder = f"pharmvar-{version}"
super().__init__(genes, filetype, db_folder=pharmvar_folder, version=version)
__iter__(self) -> Iterable[str]
inherited
special
Source code in pyhlamsa/gene_family/cyp.py
def __iter__(self) -> Iterable[str]:
"""Iter gene name like iter(dict)"""
return iter(self.genes)
items(self) -> Iterable[tuple[str, pyhlamsa.gene.genemsa.Genemsa]]
inherited
list gene name and msa like dict.items()
Source code in pyhlamsa/gene_family/cyp.py
def items(self) -> Iterable[tuple[str, Genemsa]]:
"""list gene name and msa like dict.items()"""
return self.genes.items()
list_db_gene(self, filetype: Union[str, Iterable[str]] = []) -> list
List the gene in folder
Source code in pyhlamsa/gene_family/cyp.py
def list_db_gene(self, filetype: TypeSet = []) -> list[str]:
"""List the gene in folder"""
return sorted(os.listdir(self.db_folder))
list_genes(self) -> list
inherited
List all the gene's name in this family
Source code in pyhlamsa/gene_family/cyp.py
def list_genes(self) -> list[str]:
"""List all the gene's name in this family"""
return list(self.genes.keys())
read_db_gene(self, gene: str, filetype: Union[str, Iterable[str]] = []) -> Genemsa
Read {gene}/{gene}.haplotypes.fasta and {gene}/{gene}.haplotypes.tsv
filetype will be ignored now
Source code in pyhlamsa/gene_family/cyp.py
def read_db_gene(self, gene: str, filetype: TypeSet = []) -> Genemsa:
"""
Read `{gene}/{gene}.haplotypes.fasta` and `{gene}/{gene}.haplotypes.tsv`
`filetype` will be ignored now
"""
# read fasta
ref_seqs = {}
for seq in SeqIO.parse(
f"{self.db_folder}/{gene}/{gene}.haplotypes.fasta", "fasta"
):
# split allele name
# e.g. rs75017182, rs56038477 PV01077 NG_008807.2 PharmVar Version:5.1.10
for name in seq.description.replace(", ", ",").split(" ")[0].split(","):
ref_seqs[name.strip()] = str(seq.seq)
self.logger.debug(f"Read sequence {ref_seqs.keys()}")
# read tsv
# split by '\t' and ignore header
var_text = open(glob(f"{self.db_folder}/{gene}/RefSeqGene/*.haplotypes.tsv")[0])
table = [i.strip().split("\t") for i in var_text if not i.startswith("#")][1:]
# Get Reference sequence
reference_seq = None
reference_name = None
for i in table:
if i[0] not in ref_seqs:
continue
if i[3] == "REFERENCE" and ref_seqs[i[0]][0]:
if reference_seq:
assert reference_seq == ref_seqs[i[0]]
continue
reference_name = i[0]
reference_seq = ref_seqs[reference_name]
if not reference_seq:
# hard-coded for DPYD, because it's reference `NG_008807.2` is missing
if gene == "DPYD":
reference_name = "rs111858276"
reference_seq = ref_seqs[reference_name]
reference_seq = (
reference_seq[: 376460 - 1] + "A" + reference_seq[376460:]
)
else:
raise ValueError(f"Not reference found in {gene}")
assert reference_name is not None
# Fill with Reference and reference is the first one
alleles = {reference_name: ""}
alleles.update(
{allele_name: reference_seq for allele_name in set(i[0] for i in table)}
)
self.logger.debug(f"Read vcf {alleles.keys()}")
# Remove duplciate
# in 5.1.10, there exist two same row
# CYP2D6*149 CYP2D6 rs59421388 NG_008376.4 8203 8203 G A substitution
# CYP2D6*149 CYP2D6 rs59421388 NG_008376.4 8203 8203 G A substitution
table_unique1 = filter(lambda i: i[3] != "REFERENCE", table)
table_unique2 = cast(tuple, map(tuple, table_unique1)) # type: ignore
table_unique = cast(list, map(list, set(list(table_unique2)))) # type: ignore
# Reconstruct msa from VCF variant
# VCF type: substitution and deleteion, insertion
# Table Header: ['Haplotype Name', 'Gene', 'rsID', 'ReferenceSequence',
# 'Variant Start', 'Variant Stop', 'Reference Allele', 'Variant Allele', 'Type']
# Insertion will move the index, so insert from last position first
for i in sorted(table_unique, key=lambda i: -int(i[4])):
if i[8] == "substitution":
pos = int(i[4]) - 1
end = pos + 1
elif i[8] == "deletion":
pos = int(i[4]) - 1
end = pos + len(i[6])
i[7] = "-" * len(i[6])
elif i[8] == "insertion":
# hard coded bugs
if i[0].startswith("CYP2A6*31"):
# CYP2A6*31.001 4530 4552 CCCCTTCCTGAGACCCTTAACCC - deletion
# CYP2A6*31.001 4530 4531 - AATCCATATGTGGAATCTG insertion
continue
if i[0].startswith("CYP2A6*27"):
# CYP2A6*27.001 7183 7184 - A insertion
continue
pos = int(i[4])
end = int(i[4]) + len(i[7])
i[6] = "-" * len(i[7])
# check the gap is already inserted by others
# if false: insert '-' for all alleles
# else: replace the '-' with insertion seq
if set(alleles[i[0]][pos:end]) != set("-"):
for allele_name in alleles:
alleles[allele_name] = (
alleles[allele_name][:pos]
+ i[6]
+ alleles[allele_name][pos:]
)
else:
raise ValueError(f"Unknown Type {i[8]}")
assert alleles[i[0]][pos:end] == i[6]
alleles[i[0]] = alleles[i[0]][:pos] + i[7] + alleles[i[0]][end:]
# split allele name (alleles.key() will change)
# e.g. rs75017182, rs56038477 DPYD rs75017182 NG_008807.2 346167
for allele_name in list(alleles.keys()):
if ", " in allele_name:
for an in allele_name.split(","):
alleles[an.strip()] = alleles[allele_name]
del alleles[allele_name]
# double check
length = None
for allele_name in alleles:
if length is None:
length = len(alleles[allele_name])
else:
assert length == len(alleles[allele_name])
if alleles[allele_name].replace("-", "") != ref_seqs[allele_name]:
raise ValueError(f"{allele_name} is not same as reference")
assert length
msa = Genemsa(gene)
msa.alleles = alleles
msa.blocks = [BlockInfo(length=length)]
return msa.assume_label("other")